Perfluorooctane sulfonate (PFOS) and calcium channel downstream signaling molecules
Yu
Wang
and
Yihe
Jin
*
Department of Environmental Science and Technology, Dalian University of Technology, Linggong Road 2, Dalian 116024, P. R. China. E-mail: jinyihe@dlut.edu.cn; Fax: +86-411-8470 8084; Tel: +86-411-8470 8084
First published on 18th May 2012
Abstract
As a neurotoxic environmental contaminant, perfluorooctane sulfonate (PFOS) has made a great impact on brain tissue. Neurotoxicity studies have shown that PFOS can cause abnormally high levels of calcium concentration in the hippocampus, thus inhibiting the growth of synapses, elevating the glutamate level in the nervous system, influencing behavioural response, and destroying the central nervous system, but the mechanism has not been explored by in-depth discussion. As an important second messenger in nerve cells, calcium ions mediate the release of neurotransmitters, the formation of nerve spinous process and growth through the transmission of information, and are an essential requirement for maintaining nerve physiological functions. In this paper, calcium signal transduction pathways in hippocampus cells are systematically introduced, which can help to clarify the neurotoxic mechanism of PFOS, and provide further scientific basis for learning and memory ability.
1. Calcium homeostasis and calcium signaling transduction pathway
In the central nervous system (CNS), the hippocampus is one of the major components of the brain, and is a region which is associated with a variety of neurological diseases.1 It plays important roles in the consolidation of information from short-term memory to long-term memory. The form of neural plasticity known as long-term potentiation (LTP) was first discovered to occur in the hippocampus in rabbits.2 Therefore, hippocampus neuronal plasticity is commonly used in learning and memory changes in many disease research.3 The hippocampus has frequently been used as a model system for studying neurotoxicology.4–9Calcium signaling is arguably the most important signal pathway in nerve cells, and is an important indicator to evaluate the status of the cell’s physiological state.10–13 As the second messengers of cells, calcium transfers the extracellular signal to the intracellular parts, and is involved in cell regulation in a variety of ways. Calcium has been shown to participate in a variety of biological effects, including cell proliferation, differentiation, apoptosis, and other important functions.14 Cellular calcium is found mainly in the nucleus, mitochondria, endoplasmic reticulum and plasma membrane. These organella are also called calcium stores. The generation of calcium messenger is not a result of enzyme reaction, but the result of its movement. When stimuli reach the cell surface, a small amount of extracellular calcium ions are released into the cell, resulting in an increased concentration of cytosolic calcium ions, and generation of calcium signals. The calcium concentration in cellular calcium stores is several times higher than in cytoplasm; when the cells are stimulated, the stores release calcium ions into the cytosol and they are thus another way to generate calcium signals. The important role of calcium ions in hippocampal cells is to transmit the nerve signals and to promote the secretion of neurotransmitters.15 When cytosolic calcium concentration increases, high affinity binding proteins or enzymes are activated, causing a cell response. Intracellular free calcium concentration is the key in the regulation of information transmission process.16
Cells have a comprehensive monitoring system to maintain calcium homeostasis balance. There are many proteins which in the cell membrane or organelle membrane allow extracellular calcium into the cell, called calcium channels. Calcium influx can also help calcium release from calcium stores to the cytoplasm, which is calcium release. The calcium homeostasis imbalance will lead to cell damage or death and a number of important physiological responses, including cell proliferation, division, movement, secretion, morphogenesis, energy metabolism, oxygen metabolism, and sugar metabolism. Exogenous calcium influx is primarily through plasma membrane calcium ion channels; ion channels are open or closed by conformational changes to control the flow of calcium ions.11 Endogenous calcium regulation refers primarily to the cytoplasm of certain organelles, such as endoplasmic reticulum and mitochondria, and can be induced by calcium release mechanism to regulate the cytosolic calcium ion concentration. Many toxic substances in the environment will destroy cell calcium homeostasis, resulting in extracellular calcium influx and cellular calcium store release, producing cell calcium overload, inhibiting mitochondrial ATP synthesis, causing cell damage, aging and even death.17
The mechanisms responsible for regulating the influx of external calcium are well established. Many signaling pathways are sensitive to Ca2+ (Fig. 1). For example, voltage-operated channels are used to trigger the release of neurotransmitter at synaptic junctions and they contributed to dendritic action potentials. In addition, neurotransmitters can induce an influx of calcium using receptor-operated channels such as the N-methyl-D-aspartate receptors (NMDAR).11,18 Ca2+ can activate calmodulin kinase (CaMK), which activates nitric oxide synthase (NOs), prompting the release of the neurotransmitter. Ca2+ can also activate protein kinase C (PKC), to promote Ca2+ influx, enhance the postsynaptic potential, and then maintain a longer period of LTP. Neuronal development, differentiation and synaptic plasticity are closely related to cyclic adenosine 3′,5′-monophosphate (cAMP)-response element binding protein (CREB),19,20 and the regulatory pathway including cAMP-protein kinase A (PKA), Ras-Raf-MEK-ERK (MAPK) and so on. The activation of the Ras-Raf-MAPK pathway must be dependent on intracellular Ca2+ concentration. Abnormally increased intracellular calcium concentration in nerve cells caused by environmental pollutants leading to cell signaling system abnormalities, as well as the impact of synaptic plasticity, is a focus of the field of neurotoxicology.
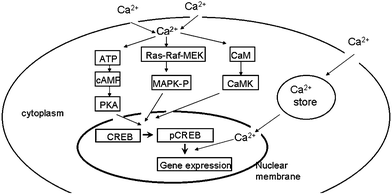 | ||
| Fig. 1 Molecules in calcium signaling. ATP, adenosine triphosphate; CaM, calmodulin; CaMK, calmodulin kinase; cAMP, cyclic adenosine 3′,5′-monophosphate; CREB, cAMP response element binding protein; MAPK, mitogen-activated protein kinase; PKA, protein kinase A; and Ras-Raf-MEK, mitogen-activated protein kinase/ERK kinase. | ||
2. Perfluorooctane sulfonate (PFOS) and calcium channel downstream signaling molecules
Recently, perfluorooctane sulfonate (PFOS) was reported to be ubiquitously detected in the environment, such as soil, water, air and other environmental media, as well as in human serum,21–26 and is well known to have toxic effects on the physiological functions of various tissues. It has lipid-repellent and water-repellent characteristics, which makes it useful for clothing fabrics, carpets, food packaging, lubricants, surfactants and fire extinguishers.27,28 In May 2009, the Stockholm Convention listed PFOS as a persistent organic pollutant.A lot of studies showed that PFOS has reproductive and developmental toxicity, immune toxicity, liver toxicity, endocrine disruptor activity and other toxicity. These studies have shown that PFOS is a class of environmental pollutant with versatile toxicities.29–36 Studies have shown that the nervous system may be one of the target organs of PFOS. Neurotoxicity studies have shown that PFOS can inhibit the growth of synapses, elevate the glutamate level in the nervous system, influence behavioral response, and destroy the central nervous system.37,38 The developing nervous system is thought to be particularly sensitive to PFOS by the fact that PFOS can cross the blood–brain and placental barriers. Although the fate, transport, distribution and bioaccumulation of PFOS have been documented, its potential neurotoxicity remains largely unknown.
An increasing number of proteins have also been identified that are likely to contribute to the structural organization of intracellular Ca2+ stores. Calcium-binding protein as the media of calcium signaling plays a very important role in cell signaling pathway, especially in the CNS in physiological and pathological processes.39
In the CNS, many responses raise the level of intracellular calcium by calmodulin protein (CaM) kinase-mediated signaling pathway. CaM is a ubiquitous calcium-dependent protein, regulating many processes in eukaryotes, such as cytoskeletal organization, vesicle transport, and the occurrence of mitosis. At the same time, CaM is also involved in the regulation of calcium and activation of certain enzymes. Calcium pump requires the activation of CaM. Both too much and too little expression of CaM will result in calcium metabolism disorders, which leads to cell damage. CaMK activity is regulated by calcium–CaM binding and phosphorylation. It regulates well synaptic plasticity, learning and memory. NMDAR, which depends on NMDAR subtype-2B (NR2B) during the early-postnatal period in hippocampus, mediated calcium dependent signaling works oppositely on neuronal function and survival.18,40,41 The expression of NR2B is usually necessary for learning and memory, developing and maintaining cellular connections.4 The multifunctional enzyme, calmodulin-dependent protein kinase (CaMK II) is required for LTP and plays a role in neuronal survival.42,43 CaMK II is an important signaling molecule on the Ca2+ signal transduction pathway, and its overexpression can inhibit axonal growth, affecting the normal physiological role of the nervous system.7 After new born offspring’s exposure to PFOS, CaMKII expression increased in the hippocampus and cortex. The expression of CaMK IIα and pCREB significantly increases in the cortex and hippocampus of rats after treatment with PFOS at dosages of 1.7, 5.0, and 15.0 mg L−1 by drinking. This also led to the increased expression of c-fos and c-jun in rats’ brains.43 PFOS given directly to the neonatal mice on postnatal day 10 can significantly increase the levels of CaMKII, GAP-43, and synaptophysin in the hippocampus of the neonatal mouse.16 This indicates that the neurotoxic effect of PFOS is partly mediated by the Ca2+-dependent molecules in calcium signaling.
Cross-foster model has been built to evaluate the possible mechanisms of the developmental neurotoxicity of PFOS. Exposure to PFOS during the critical period of development of the brain may have neurotoxic effects on the CNS by mediating the molecules of calcium signaling pathway. The result shows that the expression of calcium-related signaling molecules, such as NR2B, CaM, CaMK II and CREB was changed in the PFOS exposure group at postnatal days.44,45 As a target of calcium signaling, much research has shown that dysregulation of the CREB gene leads to neurodegeneration.40,46
Ca2+-dependent mechanisms are important in regulating synaptic transmission.47 In recent years, more studies have shown that the Wnt signaling plays a key role modulating cell differentiation or proliferation states.48 Members of the vertebrate Wnt family have been subdivided into two functional classes according to their biological activities.49 In one type, Wnt/β-catenin is critical in the development of the nervous system, including the cortex model building and synapse formation. In another type, activation of the Wnt/Ca2+ pathway may result in intracellular Ca2+ release, and activation of CaMK II and PKC, which are rarely involved in the PFOS neurotoxicity study.
Studies have shown that an abnormally increased concentration of intracellular free calcium ions in nerve cells is the common pathway of nerve tissue damage and neuronal apoptosis.46 Contrary to the other intracellular messengers, calcium initiates a stereotyped injury response not only through the dependent protein kinase, but also through a wide range of target molecules. Many different types of calcium signaling pathway increase the diversity and complexity of research.
3. Research into PFOS effects on calcium signal pathway in nervous system
Currently, toxic effects on the CNS due to calcium disorder by entry of the pollutant into the brain, mainly through channels on cell membrane, are being reported. However, little is known about the abnormal calcium increase of calcium store evoked by PFOS.Pollutants affect the intracellular calcium concentration in the cell mainly through the regulation of calcium homeostasis in two ways: extracellular calcium influx and intracellular calcium release. Calcium signaling pathway, such as calcium homeostasis and downstream molecules is easily disrupted by many chemicals.50 Calcium overload may activate downstream signaling molecules, leading to toxic effects in various cells and tissues.
PFOS induces the increase in hippocampal cell [Ca2+]i by influencing the composition of the cell surface (Fig. 2). PFOS-induced activation of cell surface receptors triggers potential changes both inside and outside the cell membrane, and activates the L-type voltage gated calcium channels, resulting in calcium influx. Harada et al.51 investigated the effects of PFOS on action potentials and L-type Ca2+ currents (ICaL) in isolated guinea-pig ventricular myocytes, finding that PFOS may change membrane surface potential, thereby eliciting general effects on calcium channels. This mechanism was also confirmed by Kawamoto et al.52 in a study with the parade of paramecium and the membrane surface potential associated with PFOS.
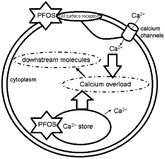 | ||
| Fig. 2 Diagram of disordered calcium induced by PFOS. | ||
In vitro models used in the present study showed that PFOS could be accumulated in neuronal cells and induce a calcium increase not only extracellularly, but also via liberation of intracellular calcium stores (Fig. 2). Primary cultures of rat hippocampal neurons indicate that PFOS can accumulate in cultured neurons and elevate calcium concentrations via release of intracellular calcium stores. Calcium released from intracellular stores may partially account for the perturbation of calcium homeostasis caused by PFOS.53 Liao et al.54 also pointed out that intracellular calcium stores participate in PFOS induced [Ca2+]i in hippocampal cells. This study showed that PFOS enhanced inward Ca2+ currents and increased intracellular Ca2+ in cultured neurons. Moreover, prolonged treatment with PFOS moderately inhibited neurite growth and dramatically suppressed synaptogenesis in cultured neurons in a nifedipine-sensitive manner. Also, PFOS-induced activation of cell surface receptors, primarily by changing the concentration of amino acids, activating the glutamate receptor, then activating NMDAR regulation of ligand-gated calcium channels, causes the calcium influx. In vitro investigation of the mechanism found that PFOS can change hippocampal cell L-type calcium channels, causing Ca2+ influx, and the occurrence of synaptic inhibition, resulting in neurotoxicity.55
After 60 days’ exposure to PFOS, hippocampal cells’ [Ca2+]i were increased compared with the control group, and there was a significant positive correlation between [Ca2+]i and PFOS concentration in serum and brain.56 The pregnant rats were exposed to PFOS with doses of 0, 7.2 or 14.4 mg kg−1 in food from pregnancy day 1 to postnatal day 35. As the days increased, [Ca2+]i in the PFOS group increased significantly.57 The results show that [Ca2+]i increased in the hippocampus of the rats, not only in the adults but also in pups exposed to PFOS with different times, which might have a neurotoxic effect on the CNS. PFOS-induced intracellular [Ca2+]i increase may lead to nervous system calcium signal transduction pathways being affected.
Taken together, these findings indicated that PFOS disturbs the neuronal physiological processes, which reveals the damage of this pollutant to nerve system and will be helpful for further exploration of its underlying mechanism. However, little is known about the potential mechanism of PFOS exposure on the CNS, and more in vivo research should be done to explore the neurotoxic mechanism of PFOS.
4. Conclusion
The CNS is a target organ of PFOS, and PFOS can pass through the blood–brain barrier, becoming enriched in brain tissue, and impacting on the nervous system.37 Johansson et al.7 reported that exposure to 1.4 or 2.1 mmol kg−1 body weight of PFOS at the age of 10 days can induce adult behavioral defects in mice, affect the cholinergic system, confirming that PFOS has potential toxic effects on the development of the nervous system. The neurotoxicity mechanism is relatively complex, and research data are still lacking. Intracellular calcium is an important cellular material basis of physiological function, which can initiate and regulate responses of central nervous tissues to injury. Damaged or stimulated cells are often accompanied by an anomalous intracellular calcium ion concentration, which results in cell calcium homeostasis imbalance, more serious injury and even death.At present, the research data on the impact of PFOS on downstream signaling molecules and the regulation mechanism of calcium ion concentration is still very limited. Existing research on the mechanism of neurotoxicity from PFOS is mainly on calcium signal transduction pathways but there is no in-depth discussion on the incidence and transmission of calcium signaling molecules. Calcium homeostasis imbalance can cause LTP, LTD damage, and inhibition of synaptic transmission occurrence, resulting in a decline in learning and memory. PFOS induced learning and memory impairment is connected with intracellular calcium concentration. However, the mechanism of the effect of PFOS-induced abnormal elevation of intracellular calcium levels in the hippocampus on LTP and LTD has not yet been discovered. As important cell signaling ambassadors, calcium ions are a key component of cell signaling, with changes in the concentration of calcium ions resulting in cellular signal transduction system abnormalities, which leads to changes in the relevant levels of protein and gene expression in the nervous system. Calcium ions affect neurotransmitter release, excitability, formation of the nerve spinous process and growth of intracellular calcium signaling pathway through the influence of calcium-dependent protein phosphorylation and ion channel activity, and are an essential requirement to maintain nerve physiological functions. Currently, the available research on the calcium signal transduction pathways is very limited, and the calcium signaling pathway is a very complex process.
In summary, these findings contribute to the understanding of how PFOS disrupts the function of neurons at different levels, which needs to be further explored.
Acknowledgements
The authors thank the National Natural Science Foundation of China (grant number 30771772) for funding during the study.Notes and references
- H. Ye, S. Jalini, S. Mylvaganam and P. Carlen, Neurobiol. Aging, 2010, 31, 591 CrossRef CAS.
- T. V. P. Bliss and T. Lømo, J. Physiol., 1973, 232, 331 CAS.
- J. Yang, Q. Liu, L. Zhang, S. Wu, M. Qi, S. Lu, Q. Xi and Y. Cai, Toxicol. Lett., 2009, 190, 208 CrossRef CAS.
- T. V. P. Bliss and G. L. Collingridge, Nature, 1993, 361, 31 CrossRef CAS.
- C. Liao, L. Cui, Q. Zhou, S. Duan and G. Jiang, Environ. Toxicol. Pharmacol., 2009, 27, 338 CrossRef CAS.
- D. M. Bannerman, J. N. P. Rawlins, S. B. McHugh, R. M. J. Deacon, B. K. Yee, T. Bastb, W. N. Zhang, H. H. J. Pothuizen and J. Feldon, Neurosci. Biobehav. Res., 2004, 28, 273 CrossRef CAS.
- N. Johansson, A. Fredriksson and P. Eriksson, NeuroToxicology, 2008, 29, 160 CrossRef CAS.
- J. L. Butenhoff, D. J. Ehresman, S. Chang, G. A. Parker and D. G. Stump, Reprod. Toxicol., 2009, 27, 319 CrossRef CAS.
- P. Zhao, Y. L. Huang and J. S. Cheng, Neurosci. Lett., 1999, 268, 25 CrossRef CAS.
- V. Konieczny, M. V. Keebler and C. W. Taylor, Semin. Cell Dev. Biol., 2012, 23, 172 CrossRef CAS.
- M. J. Berridge, Neuron, 1998, 21, 13 CrossRef CAS.
- V. Sorrentino and R. Rizzuto, Trends Pharmacol. Sci., 2001, 22, 459 CrossRef CAS.
- K. Kleszczyński, P. Stepnowski and A. C. Składanowski, Toxicol. Appl. Pharmacol., 2011, 251, 163 CrossRef.
- Y. Komori, M. Tanaka, M. Kuba, M. Ishii, M. Abe, N. Kitamura, A. Verkhratsky, I. Shibuya and G. Dayanithi, Cell Calcium, 2010, 48, 324 CrossRef CAS.
- A. Meir, S. Ginsburg, A. Butkevich, S. G. Kachalsky, I. Kaiserman, R. Ahdut, S. Demirgoren and R. Rahamimoff, Physiol. Rev., 1999, 79, 1019 CAS.
- N. Johansson, P. Eriksson and H. Viberg, Toxicol. Sci., 2009, 108, 412 CrossRef CAS.
- Y. Gong, Y. Chai, J. H. Ding, X. L. Sun and G. Hu, Neurosci. Lett., 2011, 488, 76 CrossRef CAS.
- P. Vanhoutte and H. Bading, Curr. Opin. Neurobiol., 2003, 13, 366 CrossRef CAS.
- B. E. Lonze and D. D. Ginty, Neuron, 2002, 35, 605 CrossRef CAS.
- C. Sala, S. R. Correia and M. Sheng, J. Neurosci., 2000, 20, 3529 CAS.
- J. P. Giesy and K. Kannan, Environ. Sci. Technol., 2001, 35, 1339 CrossRef CAS.
- J. D. Johnson, S. J. Gibson and R. E. Ober, Fundam. Appl. Toxicol., 1984, 4, 972 CrossRef CAS.
- Y. H. Jin, W. Liu, I. Satob, S. F. Nakayama, K. Sasaki, N. Saito and S. Tsuda, Chemosphere, 2009, 77, 605 CrossRef CAS.
- N. P. H. Lein, S. Fujii, S. Tanaka, M. Nozoe and H. Tanaka, Desalination, 2008, 226, 338 CrossRef CAS.
- G. W. Olsen, T. R. Church, J. P. Miller, J. M. Burris, K. J. Hansen, J. K. Lundberg, J. B. Armitage, R. M. Herron, Z. Medhdizadehkashi, J. B. Nobiletti, E. M. O'Neill, J. H. Mandel and L. R. Zobel, Environ. Health Perspect., 2003, 111, 1892 CrossRef CAS.
- X. Ye, M. J. Strynar, S. F. Nakayama, J. Varns, L. Helfant, J. Lazorchak and A. B. Lindstrom, Environ. Pollut., 2008, 156, 1227 CrossRef CAS.
- M. Houde, J. W. Martin, R. J. Letcher, K. R. Solomon and D. C. G. Muir, Sci. Technol., 2006, 40, 3463 CrossRef CAS.
- B. D. Key, R. D. Howell and C. S. Criddle, Environ. Sci. Technol., 1997, 31, 2445 CrossRef CAS.
- D. J. Luebker, M. T. Case, R. G. York, J. A. Moore, K. J. Hansen and J. L. Butenhoff, Toxicology, 2005, 215, 126 CrossRef CAS.
- F. Wang, W. Liu, Y. Jin, J. Dai, W. Yu, X. Liu and L. Liu, Environ. Sci. Technol., 2010, 44, 1847 CrossRef CAS.
- U. N. Joensen, R. Bossi, H. Leffers, A. A. Jensen, N. E. Skakkebæk and N. Jørgensen, Environ. Health Perspect., 2009, 117, 923 CAS.
- C. Lau, J. R. Thibodeaux, R. G. Hanson, J. M. Rogers, B. E. Grey, M. E. Stanton, J. L. Butenhoff and L. A. Stevenson, Toxicol. Sci., 2003, 74, 382 CrossRef CAS.
- A. M. Seacat, P. J. Thomford, K. J. Hansen, L. A. Clemen, S. R. Eldridge, C. R. Elcombe and J. L. Butenhoff, Toxicology, 2003, 183, 117 CrossRef CAS.
- J. R. Thibodeaux, R. G. Hanson, J. M. Rogers, B. E. Grey, B. D. Barbee, J. H. Richards, J. L. Butenhoff, L. A. Stevenson and C. Lau, Toxicol. Sci., 2003, 74, 369 CrossRef CAS.
- W. Völkel, O. Genzel-Boroviczény, H. Demmelmair, C. Gebauer, B. Koletzko, D. Twardella, U. Raab and H. Fromme, Int. J. Hyg. Environ. Health, 2008, 211, 440 CrossRef.
- D. Yahia, C. Tsukuba, M. Yoshida, I. Sato and S. Tsuda, J. Toxicol. Sci., 2008, 33, 219 CrossRef CAS.
- M. E. Austin, B. S. Kasturi, M. Barber, K. Kannan, P. S. MohanKumar and S. M. J. MohanKumar, Environ. Health Perspect., 2003, 111, 1485 CrossRef CAS.
- S. Fuentes, P. Vicens, M. T. Colomina and J. L. Domingo, Toxicology, 2007, 242, 123 CrossRef CAS.
- A. Verkhratsky, R. K. Orkand and H. Kettenmann, Physiol. Rev., 1998, 78, 100 Search PubMed.
- G. E. Hardingham and H. Bading, Trends Neurosci., 2003, 26, 81 CrossRef CAS.
- T. Yamakura and K. Shimoji, Prog. Neurobiol., 1999, 59, 279 CrossRef CAS.
- Z. Zuo, J. Cai, X. Wang, B. Li, C. Wang and Y. Chen, Aquat. Toxicol., 2009, 92, 44 CrossRef CAS.
- X. Liu, W. Liu, Y. Jin, W. Yu, L. Liu and H. Yu, Arch. Toxicol., 2010, 84, 471 CrossRef CAS.
- H. Zeng, L. Zhang, Y. Li, Y. Wang, W. Xia, Y. Lin, J. Wei and S. Xu, NeuroToxicology, 2011, 32, 130 CrossRef CAS.
- X. Liu, W. Liu, Y. Jin, W. Yu, F. Wang and L. Liu, Arch. Toxicol., 2010, 84, 71 CrossRef CAS.
- B. E. Lonze, A. Riccio, S. Cohen and D. D. Ginty, Neuron, 2002, 34, 371 CrossRef CAS.
- P. T. Kelly, R. L. MacKinnon II, R. V. Dietz, B. J. Maher and J. Wang, Mol. Brain Res., 2005, 135, 232 CrossRef CAS.
- E. M. Toledo, M. Colombres and N. C. Inestrosa, Prog. Neurobiol., 2008, 86, 281 CrossRef CAS.
- M. Kühl, L. C. Sheldahl, M. Park, J. R. Miller and R. T. Moon, Trends Genet., 2000, 16, 279 CrossRef.
- S. Schäfer, U. Bickmeyer and A. Koehler, Comp. Biochem. Physiol., C: Comp. Pharmacol. Toxicol., 2009, 150, 261 CrossRef.
- K. Harada, F. Xu, K. Ono, T. Iijima and A. Koizumi, Biochem. Biophys. Res. Commun., 2005, 329, 487 CrossRef CAS.
- K. Kawamoto, Y. Nishikawa, K. Oami, Y. H. Jin, I. Sato, N. Saito and S. Tsuda, J. Toxicol. Sci., 2008, 33, 155 CrossRef CAS.
- X. Liu, Y. Jin, W. Liu, F. Wang and S. Hao, Toxicol. in Vitro, 2011, 25, 1294 CrossRef CAS.
- C. Y. Liao, X. Y. Li, B. Wu, S. M. Duan and G. B. Jiang, Environ. Sci. Technol., 2008, 42, 5335 CrossRef CAS.
- C. Y. Liao, T. Wang, L. Cui, Q. F. Zhou, S. M. Duan and G. B. Jiang, Environ. Sci. Technol., 2009, 43, 2099 CrossRef CAS.
- X. Liu, B. Liu and Y. Jin, China Environ. Sci., 2010, 30, 110 CAS (in Chinese).
- X. Liu, B. Liu and Y. Jin, Asian J. Ecotoxicol., 2011, 6, 67 CAS (in Chinese).
| This journal is © The Royal Society of Chemistry 2012 |