Aminolevulinate dehydratase (δ-ALA-D) as marker protein of intoxication with metals and other pro-oxidant situations
Joao B. T.
Rocha
*,
Rogerio A.
Saraiva
,
Solange C.
Garcia
,
Fernanda S.
Gravina
and
Cristina W.
Nogueira
*
Centro de Ciências Naturais e Exatas, Programa de Pós-Graduação em Bioquímica Toxicológica, Universidade Federal de Santa Maria, Santa Maria, RS, Brazil. E-mail: jbtrocha@yahoo.com.br; criswn@quimica.ufsm.br; Tel: +55 55 3220 9462
First published on 30th July 2012
Abstract
δ-ALA-D is a metalloenzyme that has 3 vicinal thiol/thiolate groups that coordinate with Zn(II). The proximity between the sulfhydryl groups renders δ-ALA-D extremely sensitive to oxidation by soft electrophiles, such as Pb(II), Hg(II), As(III) and organoseleno and organotellurium compounds. In fact, blood δ-ALA-D is a classical biomarker of lead exposure in humans. The inhibition of δ-ALA-D can increase the concentration of 5-aminolevulinate (δ-ALA), which is a pro-oxidant compound. δ-ALA can generate oxidative stress that can further increase δ-ALA-D inhibition. Recently, data have been obtained indicating that the δ-ALA-D could be a marker of oxidative stress in human pathologies. In summary, considering its high sensitivity to pro-oxidant situations, δ-ALA-D can be considered a universal marker of oxidative stress.
1. δ-Aminolevulinate dehydratase or porporphobilinogen synthase: an easily oxidizable Zn(II)-thiol enzyme
Porphobilinogen synthase (PBGS, E.C. 4.2.1.24) or aminolevulinate dehydratase (δ-ALA-D) is an enzyme with a widespread distribution in nature1–5 that catalyses the asymmetric condensation of two aminolevulinic acid molecules (δ-aminolevulinate; Fig. 1) to form the monopyrrole, porphobilinogen (PBG; Fig. 1). In cells, monopyrroles are the precursors for the synthesis of tetrapyrroles. Tetrapyrroles, such as heme and chlorophyll, are essential for aerobic metabolism and carbon fixation.1–9 Consequently, toxic agents or metabolites that disrupt or interfere with tetrapyrrole synthesis can have profound effects on cell metabolism.10 Accordingly, genetic δ-ALA-D deficiency is associated with hepatic porphyria in humans which can be exacerbated or precipitated by lead intoxication.11–13 Lead can also cause 5-aminolevulinic aciduria and porphyria, which are related to δ-ALA-D inhibition.12 Recent results indicate that the δ-ALA-D interacts with proteasome and, therefore, could be a physiological modulator of proteasomal activity. Metals that interfere with the δ-ALA-D activity can also change the modulation of proteasome by the δ-ALA-D.14–18 The toxicological importance of δ-ALA-D inhibition by different metals and by oxidative stress found in a myriad pathological conditions is presented in the next sections of this review. | ||
| Fig. 1 Synthesis of porphobilinogen (PBG) from two 5-aminolevulinic acid (δ-ALA) molecules. Schematic illustration of the asymmetric condensation of two molecules of 5-aminolevulinic acid (δ-ALA) to form porphobilinogen (PBG). The A-site δ-ALA (light red) becomes the half of PBG that contains the acetyl moiety and the amino nitrogen while the P-site ALA (light blue) becomes the half of PBG that contains the propionyl moiety and the pyrrole nitrogen. | ||
Mammalian δ-ALA-D is an oligomeric Zn(II)-enzyme that is fully active as an octamer1,2,5–9,19 (Fig. 2) and it exists as an equilibrium of high-activity octamers and low-activity hexamers.1,2 Mammalian δ-ALA-D has two Zn(II) binding sites per octamer and Zn(II) of one of these sites (ZnB) participates in the catalysis as a Lewis acid9,20,21 (Fig. 3).
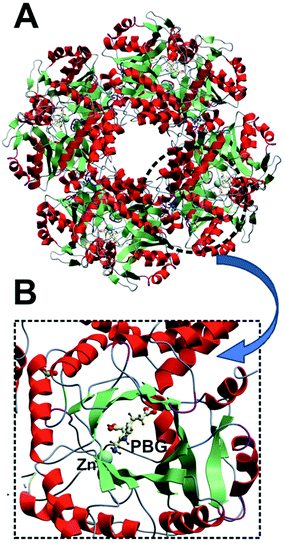 | ||
| Fig. 2 Crystal structures of active octameric (A) conformation of human δ-ALA-D (PDB entry: 1E51). According to the crystal structure, each δ-ALA-D monomer (B) presents a catalytic zinc divalent ion (Zn) and the product porphobilinogen (PBG) inside the α/β barrel active center site. The β-strands are colored in green, α-helices in red and loops in gray or purple. | ||
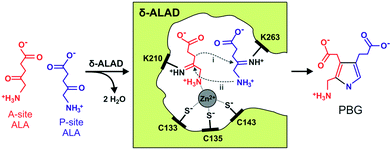 | ||
| Fig. 3 Schematic illustration of the catalytic mechanism of δ-ALA-D. The figure is based on yeast δ-ALA-D crystal structures with PDB entries 1H7O and 1OHL with the asymmetric condensation of two δ-ALA (A-site δ-ALA, in red, and P-site, in blue) moieties through two Schiff-base linkages involving the positively charged lysine residues K210 (A-site δ-ALA) and K263 (P-site δ-ALA). Zn(II) is coordinated to the thiolate groups of C133, C135 and C143 and it plays an important role in substrate binding at the A-site (by linking to the amino nitrogen of PBG) and in stabilizing intermediates and transition structures during the enzymatic reaction. | ||
One special feature of the functional structure of δ-ALA-D is the presence of vicinal cysteinyl residues in its active site23 (Fig. 3 and 4). These thiol groups are involved in the coordination of essential Zn(II) ions21–23 (Fig. 3 and 4) and the proximity between them makes the enzyme particularly sensitive to oxidation.23–25 Zn(II) is also involved in the stabilization of vicinal thiol/thiolate groups and its removal by chelating agents can accelerate enzyme autooxidation (Fig. 4).26,27 Indeed, agents that oxidize –SH groups, metals with high affinity for thiol groups or that compete with the Zn(II) binding site can inhibit δ-ALA-D.20,28–46
 | ||
| Fig. 4 Stabilization of thiol groups of δ-ALA-D by Zn(II). Removal of Zn(II) from the thiol-rich region ZnB of δ-ALA-D facilitates the oxidation of the enzyme and blocks catalysis. The preferential internal disulfide bridge after Zn(II) loss has not yet been clearly identified (three possibilities are shown in the figure). | ||
2. δ-ALA-D as a marker of metal intoxication
For a long time, blood δ-ALA-D quantification has been considered an important clinical biomarker of Pb(II) exposure. Indeed, blood δ-ALA-D can be considered an early and reliable marker of Pb(II) poisoning in workers occupationally exposed to lead.46–51 However, the modulation of δ-ALA-D by other metals, such as Hg(II), Cu(II), or Ag(II), indicates that its use to diagnose lead poisoning can be rather unspecific from the simultaneous intoxication with other metals.52 Recent results suggest that the δ-ALA-D can also be “a molecular target of pro-oxidant situations”, including here those associated with chronic-degenerative human diseases and in animal models of such diseases (details in Section 4). Thus, in addition to being a target of toxicants or metals that can directly oxidize or interact reversibly with thiol groups, the δ-ALA-D can be inhibited by an imbalance between reductive and oxidative metabolism found in different diseases.53–55Of toxicological significance, the studies of Bechara and colleagues have demonstrated that the substrate of δ-ALA-D, the 5-aminolevulinic acid or δ-ALA (Fig. 1 and 3) can exhibit pro-oxidant properties under physiologically relevant conditions.56–70 They have also demonstrated that markers of oxidative stress were altered in workers exposed to lead and postulated that this could be related to an increase in circulating 5-aminolevulinic acid (δ-ALA).61 The inhibition of δ-ALA-D in lead poisoning46–51 highlights the importance of its substrate, δ-ALA, on the molecular pathology of lead intoxication. Consequently, inhibition of δ-ALA-D by toxic agents or pathological conditions associated with oxidative stress can initiate a pro-oxidative vicious cycle that will further inhibit the δ-ALA-D and increase the concentration of potentially toxic compounds, such as δ-ALA and related metabolites.70 Supporting this hypothesis, treatment of rats with δ-ALA induced cerebral oxidative stress and δ-ALA-D inhibition. Melatonin, which has antioxidant properties, blunted the pro-oxidant effects of δ-ALA and restored δ-ALA-D to normal levels.71
As mentioned above, δ-ALA-D has been utilized as a clinical indicator of lead intoxication because the enzyme is very sensitive to in vitro and in vivo Pb(II) inhibition.72–87 However, the strong reactivity in vitro of the vicinal thiol groups of δ-ALA-D with soft electrophiles, such as Hg(II), Cd(II), Sn(II), As(III), Bi(III), In(III), Tl(III), Se(IV) and Te(IV) indicated that any electrophile could be a potential in vitro or in vivo molecular inhibitor of mammalian δ-ALA-D (Fig. 5). The in vitro and in vivo inhibition of δ-ALA-D by these soft electrophiles is presented in the following sections. Most importantly, the combined exposure to Pb(II), As(III) and Cd(II) can have interactive toxic effects increasing or decreasing δ-ALA-D inhibition, depending on the duration of exposure and the tissue considered.76 This elegant study highlights a crucial aspect that has been neglected and indicates the necessity of additional investigations to determine how these soft electrophiles interact to change or enhance their toxicological properties.
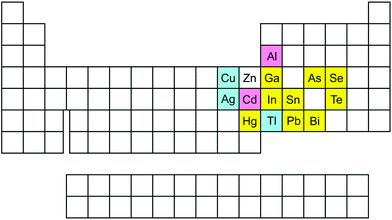 | ||
| Fig. 5 Representation of elements from the periodic table that inhibit or stimulate δ-ALA-D activity. Zn(II) is the natural ligand of δ-ALA-D. Cd(II) and Al(III) can be either inhibitors or activators of the enzyme activity; whereas Ga(III), In(III), Hg(II), RHg(II), Sn(II), Pb(II), As(III), Bi(III), Se(IV), RSe-SeR, Te(IV) and RTe-TeR (where R can be an alkyl or aryl group) are either in vitro and/or in vivo inhibitor of δ-ALA-D. Pink indicates cations that can either stimulate or inhibit δ-ALA-D activity. Blue indicates cations that have been reported to inhibit mammalian δ-ALA-D activity in vitro. Yellow indicates those that inhibit δ-ALA-D activity either in vitro or in vivo. | ||
2.1. Pb(II) in vitro
Lead is a classical inhibitor of δ-ALA-D in vitro and Zn(II) can prevent or reverse the inhibition caused by Pb(II).42,44,72–76 Treatment with Zn(II) in vitro can restore the δ-ALA-D activity following its inhibition caused by exposure to Pb(II) or Sn(II) in vivo.74 Similar results were obtained in blood of humans exposed to lead. The inhibitory action of Pb(II) is related to displacement of Zn(II) from the active site of the mammalian δ-ALA-D, rather than to a direct oxidation of the enzyme's thiol groups by Pb(II)20 (Fig. 6). Pb(II) is bulkier than Zn(II) and it, probably, cannot properly interact with amino acid residues and the substrate in the active centre of δ-ALA-D as does Zn(II) (Fig. 7A, B). Recently analysis based on molecular and computational models demonstrated that secondary bonding interactions (SBIs) and lone pair interactions profoundly influence the coordination geometry and binding affinity of Pb(II) and As(III) to peptides that simulate the environment of enzymes containing 3 thiol groups in close proximity.88 Similar phenomena could occur at the Zn(II)B binding site of δ-ALA-D. | ||
| Fig. 6 Pb(II) Competition with Zn(II) at the active site of δ-ALA-D. δ-ALA-D inhibition by Pb(II) is mediated by substitution of Zn(II) from the thiolate-rich region in the mammalian enzyme.20 | ||
 | ||
| Fig. 7 Crystal structures of δ-ALA-D complexed with Zn(II) (A); Pb(II) (B) and Hg(II). Data are from yeast δ-ALA-D (A, PDB entry 1AW5), Pb(II) (B, PDB entry 1QNV) and Hg(II) (C, PDB entry 1QML). Pb(II) and Hg(II) can replace the enzyme's catalytic Zn(II) bound by three conserved cysteine side chains (C133, C135 and C143). Red dotted lines denote metal interactions (with distances ≤ 3.5 Å). The metal ions are shown in gray or blue scalled spheres. All the amino acid residues which are involved in molecular metal interaction are shown in ball-and-stick drawings. For clarity, hydrogen atoms are hidden. Atoms in sticks are colored as follows: carbon gray, oxygen red, nitrogen dark blue and sulfur yellow. | ||
2.2. Pb(II) in vivo
Lead is a widespread toxicant that causes a variety of toxic effects in adults89 and is extremely neurotoxic to the developing brain90 causing long-lasting learning deficits in children.91 The molecular mechanisms triggering lead toxicity involve the disruption of a variety of target proteins,90–94 including δ-ALA-D. In fact, δ-ALA-D is an important blood lead binding protein94 and it has been demonstrated that in erythrocytes δ-ALA-D is the primary target of Pb(II) instead of hemoglobin.94 In contrast to blood, little is known about the relative affinity and binding of Pb(II) to the δ-ALA-D in comparison to other target proteins in soft tissues of mammals. The lack of knowledge on this subject is related to technical limitations since soft tissues contain several proteins that can bind Pb(II) and accurate information about the concentration of these targets is not available. However, from a toxicological point of view, it would be important to determine the in vitro affinity of Pb(II) to known target proteins to give support to computational studies, which could give us a rough calculation of Pb(II) distribution within living cells. Such demanding and interdisciplinary studies could fill many gaps remaining in our knowledge about the interaction of Pb(II) and other toxic metals with specific target proteins.The inhibition of δ-ALA-D by lead in vivo can be reversed by addition of Zn(II) and/or dithiothreitol (DTT) in vitro, indicating that the binding of Pb(II) in vivo is reversible. However, the strength of that inhibition is sufficient to trigger an increase in δ-ALA-D expression after exposure of rats to Pb(II).95–97 The stimulation of δ-ALA-D synthesis after Pb(II) intoxication was interpreted as a compensatory response to the enzyme inhibition. We have observed that a short-term exposure of suckling rats to high doses of Pb(II) caused an increase in blood δ-ALA-D activity35 and an increase in the reactivation index by dithiothreitol (DTT) + Zn(II) of two times,98 corroborating the findings of Fujita and collaborators.95–97
In contrast to rats, workers from battery manufacturing plants exposed to lead exhibited an increase in δ-ALA-D gene methylation and a decrease in δ-ALA-D transcription. Most importantly, the increase in δ-ALA-D gene methylation was associated with an increased risk of lead intoxication.99 These discrepancies between rats and humans can be related to species, duration and levels of lead exposure. The limited number of studies on this subject clearly indicates the necessity of more detailed scrutiny, particularly in view of the fact that δ-ALA-D gene methylation seems to be associated with an increased risk of lead toxicity in humans.
Of particular toxicological significance, Pb(II) exposure can be associated with increased oxidative stress and decreased δ-ALA-D activity.77,82–85 For instance, children with aplastic anemia or suffering from neurological disorders (such as cerebral palsy, seizures, and encephalopathy) exhibit higher levels of blood lead, elevated thiobarbituric reactive species (TBARS), and lower δ-ALA-D activity and GSH levels than healthy subjects, indicating that part of the pro-oxidant effect of lead can be mediated by δ-ALA-D inhibition.86,87 Similar results were obtained in urban adolescents with blood levels above10 μg dL−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 80 and in lead-exposed battery plant workers.81
80 and in lead-exposed battery plant workers.81
The now well-established role of oxidative stress in Pb(II) intoxication highlights the early proposals by Bechara and colleagues56–70 and strongly suggests that δ-ALA-D inhibition contributes to Pb(II) toxicity by either disrupting the heme biosynthesis pathway and/or by increasing the concentration of the potential pro-oxidant δ-ALA.
2.3. Cd(II) in vitro
Cadmium (Cd) is extremely toxic to living cells and an important environmental and occupational pollutant that can promote human diseases (e.g., cancer and renal diseases).100–103 The molecular mechanism of Cd(II) toxicity is not completely understood but it may involve disruption of Zn(II) homeostasis,104 which can in turn modify the activity of Zn(II) enzymes, such as the mammalian δ-ALA-D.105 Furthermore, Cd(II) can also stimulate the production of reactive oxygen species (ROS), either directly or indirectly via inhibition of antioxidant activities.101,103 Accordingly, Cd(II)-induced inhibition of testicular δ-ALA-D and increase in oxidative stress are reversed by the antioxidant ebselen.106In contrast to Pb(II), Cd(II) can replace Zn(II) in the active site of mammalian δ-ALA-D and promote the catalysis of PBG synthesis in vitro.75,104–109 At neutral pH, the kinetic properties of mammalian Cd(II)-ALA-D are similar to those observed with the natural ligand Zn(II).108 Davis and Avram75 demonstrated that Cd(II) is a more potent activator of human erythrocytic δ-ALA-D and reverses Pb(II)-induced enzyme inhibition more efficiently than Zn(II). However, at high concentrations Cd(II) inhibits δ-ALA-D and Zn(II) cannot re-activate Cd(II)-inhibited δ-ALA-D.75 This possibly indicates that at low concentrations Cd(II) binds to a stimulatory Zn(II) site without oxidizing or disrupting the thiol/thiolate interaction with the surrounding environment. The inhibitory effect of Cd(II) on δ-ALA-D at high concentrations can be related to its higher softness and to its stronger affinity for sulfhydryl groups than Zn(II). At low concentrations Cd(II) could replace Zn(II) and participate as Lewis acid in the catalysis, contributing to properly ionizing essential amino acid residues and substrates in the active site of the enzyme. On the other hand, at high concentrations Cd(II) could bind strongly and oxidize essential –SH group(s) involved in the coordination with Zn(II), or could oxidize –SH group(s) important for the maintenance of the native tertiary/quaternary structure of δ-ALA-D. However, the exact location of Cd(II) binding in the active centre of δ-ALA-D has not been precisely demonstrated yet and, in analogy to Pb(II), it is possible that Cd(II) can interact with the ZnA or ZnB sites of the enzyme (Fig. 8). Cd(II) can also form redox active complexes with dithiols and inhibit the mammalian ALA-D more efficiently than free Cd(II).110
 | ||
| Fig. 8 Binding of Cd(II) to the Zn(II) binding sites in δ-ALA-D. At low concentrations, Cd(II) is expected to binding to the ZnB site (C122, C124 and C132) and to substitute Zn(II) and maintain δ-ALA-D activity. At high concentrations, Cd(II) possibly oxidizes these residues or binds to additional Zn(II) binding site (ZnA) represented by C223 (adapted from ref. 20). | ||
2.4. Cd(II) in vivo
In contrast to in vitro data, which show that at low concentrations Cd(II) activates and at high concentrations it inhibits the δ-ALA-D, the results obtained following in vivo exposure are not homogeneous, probably reflecting the dose, duration of exposure and the source of the enzyme. Occupational exposure to Cd(II) has been associated with either no alterations in erythrocytic δ-ALA-D46,111 or with a positive correlation between blood Cd levels and δ-ALA-D reactivation index,112 indicating that exposure to Cd(II) can be associated with oxidation of δ-ALA-D. However, the subjects enrolled in the last study were also exposed to lead and the increase in the reactivation index could be the result of a complex interaction between Cd(II) and Pb(II) or the effect of the prevalence of Pb(II) over Cd(II).Results in the literature show that Cd(II) can cause either no modification in rodent δ-ALA-D37,113,114 or increase hepatic δ-ALA-D activity.114 The authors also demonstrated that Cd(II) decreases the inhibitory effect of Pb(II) on hepatic but not on renal enzyme in vitro. The stimulation of hepatic δ-ALA-D activity and the reduced sensitivity to Pb(II) in vitro were attributed to induction of synthesis of Zn,Cd-thioneins by Cd(II).114 In results obtained by our group, we observed an increase of the hepatic δ-ALA-D after exposure of rats to Cd(II).105 However, under our experimental conditions, the stimulation of the δ-ALA-D by Cd(II) was not associated with an increase in metallothionein synthesis, but rather with an increase of the Zn levels in the liver. Recently, Whittaker and colleagues76 demonstrated that Cd(II) could either stimulate (blood) or inhibit the δ-ALA-D (blood and kidney), depending on the length of exposure. Briefly, in vivo exposure to Cd(II) can have inhibitory or stimulatory effect on the δ-ALA-D activity; depending possibly on the concentrations reached by Cd(II) in different target tissues. The in vivo stimulatory effect of Cd(II) may be mediated by indirect induction of metallothioneins expression 114 or Zn(II) redistribution.105 Nonetheless, a direct stimulation of the δ-ALA-D by Cd(II) cannot be ruled out.
2.5. Hg(II) in vitro
Inorganic and organic mercury salts (Hg(II), CH3Hg(II), CH3CH2Hg(II)) are powerful electrophiles and have a strong affinity for soft nucleophiles. Biologically speaking, they have a strong affinity for –SH groups from target proteins.115–118 In fact, Hg(II) and CH3Hg(II) can inhibit δ-ALA-D in vitro.32,35,37,110 However, the inhibitory potency of Hg(II) or CH3Hg(II) is lower than Pb(II).32,35,119 Consequently, considering that Pb(II) has a lower affinity for –SH groups than mercurials,120 other factors in addition to affinity for thiol are possibly more important in determining the inhibition of δ-ALA-D by these metals. For instance, steric conflict, secondary bond energy interactions and lone pair interaction can strongly influence the coordination geometry and binding affinity of metals with vicinal thiols in models peptides.88Detailed structural analysis indicates that Hg(II) binds to the same region as do Zn(II) and Pb(II)21 (Fig. 7C), therefore indicating that Hg(II) can bind to the same sulfhydryl groups involved in the coordination with Zn(II). However, in view of the stronger affinity of Hg(II) for –SH groups than Pb(II) and Zn(II),120 Zn(II) is unable to reactivate the enzyme inhibited by Hg(II) in vitro as it reactivates the Pb(II)-inhibited δ-ALA-D.72–75 Furthermore, the distances of Hg(II) to cysteinyl residues in δ-ALA-D crystal are shorter than that observed with Zn(II).21 Consequently, since Hg(II) has a stronger affinity for thiol groups than Pb(II) and Zn(II), it is possible that it binds to 2 –SH groups instead of coordinating with 3 thiolates (Fig. 9).120 The interaction and binding of soft metals, such as Hg(II) and Pb(II), to thiolate-rich binding site are complex and little explored.120 In fact, factors such as the rate of metal complexation reactions with thiol-rich sites, the geometry of interactions and the competition of soft metals for the binding sites in aqueous solution have not yet been addressed in detail.120 Consequently, our knowledge about the toxicity of metals will be greatly increased with studies addressing these important factors involved in the interaction of metals with thiol-rich proteins.
 | ||
| Fig. 9 Representation of the binding of Hg(II) to ZnB site in δ-ALA-D. Although Hg(II) can coordinate with 3 thiol groups in the δ-ALA-D active centre, its high affinity for –SH groups can determine a preferential binding to only 2 thiolates (four possibilities of Hg(II) binding to ZnB are presented). | ||
CH3Hg(II) inhibits rodent δ-ALA-D in vitro;32 however, there are no structural studies indicating the exact site of CH3Hg(II) interaction with the δ-ALA-D. CH3Hg(II) may interact with the same thiol groups in δ-ALA-D that does Hg(II). In low molecular weight compounds, CH3Hg(II) preferentially binds to thiol in a 1:1 stoichiometry with a distance of about 2.4 Å.121 Consequently, CH3Hg(II) is expected to preferentially form complex(es) with a single or exceptionally with two –SH groups (Fig. 10). Indeed, in the case of the 3 cysteine residues of δ-ALA-D, the electronic structure of mercury ions (e.g. the fact that CH3Hg(II) ion will likely bind to only one thiol group, as compared to 2 thiol groups in the case of Hg(II) ion) is the key factor that dictates the stronger affinity of these groups for Hg(II), when compared with CH3Hg(II).115 Thus, CH3Hg(II) probably oxidizes one thiol group in δ-ALA-D; whereas Hg(II) oxidizes two or, exceptionally, three groups in δ-ALA-D, either by forming a crosslink between two thiol groups (R1–S–Hg–S–R2) or a strong geometrical interaction with the three vicinal thiolate groups in the active center of δ-ALA-D21 (Fig. 9 and 10). As discussed above, considering the important role of the geometry of metal binding to thiolate-rich region in proteins for their toxicity,120 it will be important to determine in detail the CH3Hg(II) and Hg(II) binding motifs in δ-ALA-D. The determination of the potential bond lengths involved in the binding of these mercurials to δ-ALA-D will perhaps indicate that the distance between the outermost Cys residues is too long for some of the bonding motifs to be feasible. Thus, the expectation is that this type of studies will contribute to increase our understanding about the toxicity of these two forms of Hg and also of related toxic soft metals.
 | ||
| Fig. 10 Hypothetical interaction of MeHg (CH3Hg(II)) with thiol groups from ZnB site in δ-ALA-D. | ||
2.6. Hg(II) in vivo
CH3Hg(II) is an important ubiquitous environmental toxicant that can be found at high concentrations in piscivorous fish.116–118,122,123 Consequently, the general population can be exposed to CH3Hg(II) via ingestion of contaminated fish. Of particular toxicological significance, there are two studies in the literature supporting the theory that the erythrocytic δ-ALA-D can be a target of CH3Hg(II). In a study carried out about 40 years ago, Schutz and Skerfving124 reported that the δ-ALA-D activity in blood of subjects exposed to CH3Hg(II) via fish consumption was lower than that of non-exposed subjects and the inhibition was proportional to the total amount of Hg in the blood. Recently, negative correlations have been observed between the erythrocytic δ-ALA-D activity and Hg levels in the blood and hair of fish-eating subjects from the Amazonian region.125 Antioxidant defense markers were negatively correlated with Hg levels, namely the antioxidant enzymes glutathione peroxidase and catalase. The authors also showed that the δ-ALA-D reactivation index was positively correlated with Hg levels and negatively correlated with antioxidant enzymes, supporting the notion that oxidative stress contributes to oxidizing the erythrocytic δ-ALA-D in Hg-exposed subjects. The study by Grotto et al.125 illustrates that the inhibitory effect of CH3Hg(II) on δ-ALA-D activity can be mediated by a direct oxidation of thiol groups by CH3Hg(II) or indirectly via oxidative stress mediated oxidation of thiol groups.Experimental studies indicate that short term exposure to high doses32 or chronic exposure to low doses of CH3Hg(II) inhibit the δ-ALA-D and increase plasma MDA levels,126 indicating a relationship between oxidative stress and δ-ALA-D inhibition. Here, it is important to point out that chronic CH3Hg(II) exposure have been reported to disrupt heme biosynthesis in rodents,127,128 which may be related to δ-ALA-D inhibition.
Human exposure to inorganic mercury (Hg(0), Hg(I) or Hg(II) can occur accidentally or occupationally under diverse situations. For instance, the use of skin-lightening creams with Hg has resulted in Hg poisoning, 129 or even the bizarre use of Hg for the treatment of head lice which caused fatal fulminate hepatic failure in a child.130 The use of Hg(0) in dental amalgams and gold mining has been also associated with Hg exposure.122,131,132 Furthermore, broken thermometers and compact fluorescent light bulbs can represent a domestic source of exposure to mercury.122
Hg(II) has a very high constant affinity for thiol groups.115,121,133 However, there are no reports in the literature indicating that exposure to Hg(II) can inhibit human δ-ALA-D. In the study by Grotto et al.,125 total Hg was determined in blood, plasma and hair. Since the subjects were from fish-eating communities, the mercury is probably derived from CH3Hg(II). Conversely, the Amazonian region has been exposed to high levels of Hg(0) used in gold mining.132 Thus, without determining the speciation of Hg, we can speculate that the results of Grotto et al.125 can be in part related to exposure to Hg(II) metabolically derived from Hg(0).
Exposure of mice and rats to high doses of Hg(II) inhibits the δ-ALA-D.35–38 However, the sensitivity of the renal enzyme is greater than the hepatic δ-ALA-D, which in turn is greater than the enzyme from the brain.35–38,134–136 These results are in agreement with the preferential distribution of Hg(II) to the kidney followed by the liver. Pre-exposure to Zn(II) decreases the inhibitory effect of Hg(II) on the rat renal and hepatic δ-ALA-D,37,38 which is not related to changes in the distribution of Hg. The protection was related to metallothionein synthesis, which chelates Hg(II).
Hg(II) exposure has been reported to cause oxidative stress and inhibition of renal δ-ALA-D activity in rodents. Administration of the antioxidant lycopene blunted the pro-oxidant effect of Hg(II) and restored δ-ALA-D activity to control levels.137 In contrast, astaxanthin, a carotenoid with antioxidant properties, prevented renal lipid peroxidation, but did not reverse Hg(II)-inhibited δ-ALA-D,138 indicating a direct inhibitory effect of Hg(II) on the renal δ-ALA-D activity.
2.7. Al(III) in vitro
Aluminum is one the most abundant element on the earth's crust and it is largely utilized by humans in industry and household utensils. Furthermore, Al(III) is also employed in medicinal formulations, water clarification and as food additive.139 Consequently, the daily exposure to aluminum can be high and the element has been implicated as a crucial factor in a variety of chronic disease.139–141 Aluminum has also been indicated as the causative factor of a microcytic hypoproliferative anemia and it can worsen anemia in patients with end-stage renal disease. Even though the mechanism of aluminum-induced anemia is not completely understood, it seems to involve the disruption of heme biosynthesis.142Al(III) has a low affinity for thiol groups.34 However, there is at least one report showing that Al(III) inhibits the δ-ALAD activity in vitro with relatively high potency (in the low μmol L−1 rage).143 In other studies, Al(III) has been shown to inhibit the δ-ALA-D only in the millimolar range.144,145 In mouse blood, low concentrations of Al(III) were shown to stimulate the δ-ALA-D.145 With purified hepatic δ-ALA-D, the inhibitory potency of Al(III) was also in the millimolar range and Zn(II) blunted the inhibitory effect of Al(III).34 The reasons for such discrepancies are unknown, but they cannot be explained based only on the enzyme sources. Further studies need to be carried out to clarify these discrepancies.
2.8. Al(III) in vivo
After in vivo exposure, Al(III) caused either no effect,146,147 stimulation144,148 or inhibition144,149 of the δ-ALA-D activity, depending on the tissue, the chemical form of Al(III) administration (e.g. Al(III)-citrate or Al(III)) and the route of administration (oral or intraperitoneal).2.9. Ga(III), In(III) and Tl(III) in vitro and in vivo
2.10. As(III) in vitro and in vivo
Arsenic (As) is a widespread pollutant and its presence in water is a serious health hazard in several parts of the world.163 Arsenic is thought to be involved in various adverse health effects, including cancer and neurodegenerative diseases.164,165 However, the mechanism of arsenic toxicity is not completely known. As(III) can interact with thiol groups of proteins, disrupting their physiological roles. Accordingly, As(III) is a weak inhibitor of the rodent δ-ALA-D in vitro.39,41 The molecular interaction of As(III) with δ-ALA-D has not yet been investigated. However, since As(III) and Pb(II) bind in a similar trigonal pyramidal coordination environments within peptidic frameworks using cysteinate ligands88 and the δ-ALA-D has 3 vicinal thiol groups; it is possible that As(III) coordinate with thiolate groups of the enzyme with a similar geometry as does Pb(II). Corroboration of this inference can be found in studies that show that concomitant exposure to As(III) and Zn(II) in vivo protects the δ-ALA-D from As(III)-induced inhibition. On the other hand, post-treatment does not protect δ-ALA-D from As(III).166,167There are several studies demonstrating that exposure of rodents to As(III) causes inhibition of the δ-ALA-D39,41,76 and increases oxidative stress.166–170 The administration of different types of antioxidant agents reversed the alterations induced by As(III).171,172 However, quercitin did not restore the δ-ALA-D following the treatment with As(III), even though quercitin blunted the As(III)-induced oxidative stress.173 This indicates that As(III) can directly oxidize the critical thiol groups of δ-ALA-D.
2.11. Sn(II) and Bi(III) in vitro and in vivo
2.12. Children’s exposure to metals
Few studies have investigated the effects of metals on δ-ALA-D activity in children, with the majority of studies focusing their attention on the effects of lead.186–189 Children seem to be much more susceptible to lead intoxication as a consequence of their hand to mouth habit, increased gastrointestinal absorption and elevated respiratory rate.189,190 Environmental pollution is the greatest source of child contamination with metals.188 Children may be directly exposed to lead in dust, water and food.191 Other sources of lead intoxication have been found in Mexican children, suggesting that the use of lead-glazed dishes and the habit of biting colored pencils increase lead levels in children.191 The consequences of this exposure are deleterious to developing subjects, leading to a higher risk of neurological damage and cancer.188 Moreover, δ-ALA-D activity has been shown to decrease after exposure to Pb, resulting in severe hematological consequences during development.192 For instance, a recent study has shown that lead induced oxidative stress and decreased δ-ALA-D activity in children with aplastic anemia.86 Thus, δ-ALA-D activity has been suggested to be a sensitive indicator of early hematological disorders related to lead exposure.78Similarly to what happens in adults, children may also have variable susceptibility to lead intoxication193 due to δ-ALA-D polymorphisms. Adult carriers of the ALAD-2 allele present higher blood lead levels and initially it was interpreted as they could be more susceptible to lead intoxication.194 However, later on, it was found that δ-ALAD-2 has higher affinity and stability for lead than δ-ALAD-1.195 Therefore, carriers of ALAD-2 allele may be less susceptible to intoxication, because of a putative protective effect of this polymorphism.196 Child carriers of ALAD-2 allele also present significantly higher blood lead levels than ALAD-1 carriers.197 Thus, they may also have a protective mechanism against Pb(II) toxicity. Interestingly, a study in Chile showed that children living in areas close to lead deposits are carriers of ALAD-2 allele more frequently than those living far away, suggesting a long-term selective defense associated with ALAD-2 polymorphism.198
The few studies cited above indicate that δ-ALA-D has not been sufficiently explored as a marker of metal exposure in children. Since children at risk of exposure are normally exposed to more than one toxic agent, it would be important to determine the activity of this enzyme in subjects at risk of exposure to different environmental toxicants.
3. δ-ALA-D as a marker of exposure to pro-oxidant compounds
3.1. Exposure to organochalcogens
Organochalcogen compounds are defined as the structures in which the chalcogen atom is directly bonded to a carbon (sp, sp2 or sp3) atom. In the following section the term organochalcogen will be used to refer specifically to organoselenium and organotellurium compounds.Understanding of the molecular toxicity of organochalcogen compounds is still scarce in the literature. Much of the present knowledge of organochalcogen toxicity emerged from inorganic selenium research. Inorganic Se compounds can oxidize thiol groups and, in the case of selenium (Se(IV)), generate reactive oxygen species (ROS) during the catalytic oxidation of thiols.199–202 These factors are believed to be the cause for toxicity of inorganic Se. To some extent, the interaction of organochalcogens with thiols has been reported to be similar to that associated with inorganic Se (for recent reviews see ref. 203–206 and references therein). The next paragraphs are intended to provide the reader with knowledge about the in vitro and in vivo inhibition of the δ-ALA-D by organochalcogens.
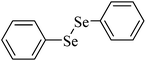
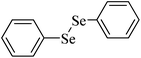
The importance of oxygen for the inhibitory effect of diphenyl diselenide and analogues was experimentally demonstrated and its removal from the medium decreased the inhibitory effect.207 From these experiments evidence emerged that molecular oxygen can oxidize the selenol intermediates back to a diselenide (Fig. 11, panel A). The inhibition of the mammalian δ-ALA-D activity by diphenyl diselenide (and analogues) was dependent on the oxidation of cysteinyl residues located within the active site of the enzyme (Fig. 11, panel A).207
 | ||
| Fig. 11 Diphenyl diselenide inhibits δ-ALA-D activity from rats (panel A) but not from plants (panel B). The importance of vicinal thiol groups for inhibition of the rat enzyme. | ||
The presence of vicinal thiols in the active center of the mammalian,208 fruit fly209 and fish210 δ-ALA-D indicates that they are crucial for the inhibitory action of diphenyl diselenide. This hypothesis was further supported by the fact that the δ-ALA-D from cucumber leaves is not sensitive to diphenyl diselenide (or its analogues).24,25,207 In plants, the B site of δ-ALA-D is characterized by the substitution of some cysteinyl residues by acidic amino acids, which makes the enzyme less susceptible to oxidation (Fig. 11, panel B).211
The use of classical reducing agents, such as DTT, to restore δ-ALA-D activity reinforces the notion that organochalcogens inhibit the enzyme by oxidizing its essential cysteinyl residues (Fig. 11, panel A).24,207,208 Despite the relatively poor thiol oxidant property of monoselenides, there is substantial evidence indicating that they can oxidize the δ-ALA-D after chemical or enzymatic transformation to their respective selenoxides.212–215Table 1 shows that selenoxides are more potent inhibitors of the rat liver δ-ALA-D than their respective parent compounds. This effect can be explained by the presence of selenium–oxygen double bond in which the pi (π) electron delocalization makes the selenium atom of selenoxides more electrophilic than that of selenides.
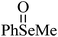

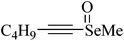
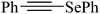
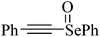
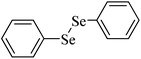
Taking into consideration the above mentioned and that the monooxygenation of selenides to their selenoxides by flavin-containing monooxygenases (FMO) has been reported in vivo,214 δ-ALA-D could be a potential target of selenides toxicity after their oxidation to selenoxides by FMO.
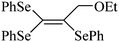
Table 1 also shows that phenylselenoacetylene at high concentrations inhibits the δ-ALA-D activity from rat liver. This inhibitory effect was dependent on the conversion of phenylselenoacetylene to diphenyl diselenide.215
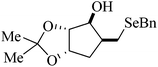
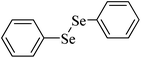
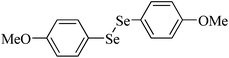
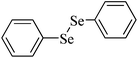
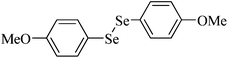
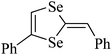
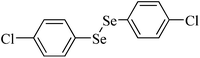
A number of organochalcogen compounds are inhibitors of the δ-ALA-D activity. Diselenides (Table 2, entries 1–4) are more effective inhibitors than the other organochalcogens shown in Table 2. The inhibitory potential of diphenyl diselenide varies depending on the source of enzyme (Table 2, entries 1, 7 and 18). The human erythrocytic δ-ALA-D is less sensitive to diphenyl diselenide than the rat liver or brain.
| Entry | Compound | Enzyme source | IC50 (μM) | Ref. |
|---|---|---|---|---|
| a IC50 values were calculated using data obtained after pre-incubation of different concentrations of organochalcogens and the source of δ-ALA-D; for details of the experimental procedure readers should refer to the references cited above. | ||||
| 1 |
|
Rat liver | 9 | 207 |
| 2 |
|
Rat liver | 2 | 207 |
| 3 |
|
Rat liver | 9 | 207 |
| 4 |
|
Rat liver | 10 | 216 |
| 5 |
|
Rat liver | >400 | 216 |
| 6 |
|
Rat liver | >200 | 216 |
| 7 |
|
Rat brain | 4.6 | 217 |
| 8 |
|
Rat brain | 6.6 | 217 |
| 9 |
|
Rat brain | 4.3 | 217 |
| 10 |
|
Rat brain | 4.3 | 217 |
| 11 |

|
Mouse brain | 171 | 218 |
| 12 |
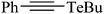
|
Mouse brain | 177 | 218 |
| 13 |

|
Mouse brain | 204 | 218 |
| 14 |
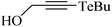
|
Mouse brain | 213 | 218 |
| 15 |
|
Human erythrocyte | >200 | 219 |
| 16 |
|
Human erythrocyte | >200 | 219 |
| 17 |
|
Human erythrocyte | >400 | 219 |
| 18 |
|
Human erythrocyte | 40 | 208 |
| 19 |
|
Human erythrocyte | 39 | 208 |
| 20 |
|
Human erythrocyte | 100 | 208 |
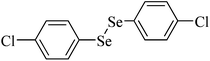
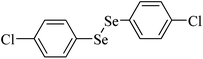
Diaryl diselenide substituted with chloro at the para position of the aromatic ring is the most effective inhibitor of the rat liver δ-ALA-D (Table 2, entry 2).
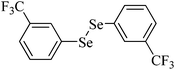
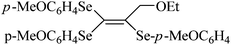
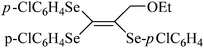
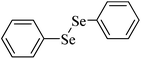
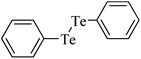
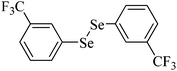
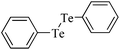
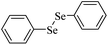
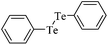
Despite the differences in the organic structure bonded to the selenium atom, carbohydrate diselenide derivatives were as potent as diphenyl diselenide in inhibiting δ-ALA-D from rat liver (Table 2, entries 1 and 4). By contrast, the IC50 values of diphenyl diselenide and its analogues were similar when the source of δ-ALA-D was rat brain, suggesting that the introduction of functional groups (trifluoromethyl, chloro or methoxyl) into the aromatic ring of diaryl diselenide does not alter its inhibitory effect (Table 2, entries 7–10). As seen on Table 2, selenofuranoses (entries 5–6)216 and tris-selenides (entries 15–17)217 are fair inhibitors of δ-ALA-D activity. It is also evident that δ-ALA-D from human erythrocytes is a potential target for diphenyl diselenide, ebselen and diphenyl ditelluride (Table 2, entries 18–20).208
The above mentioned studies provided clear evidence that δ-ALA-D from different sources can be a marker of organochalcogen exposure in vitro. Furthermore, δ-ALA-D inhibition may increase the concentration of 5-aminolevulinic acid (δ-ALA), which is a pro-oxidant molecule (Fig. 12).68–70 Extrapolating the in vitro findings, the disruption of the aerobic metabolism by inhibiting the heme biosynthesis (Fig. 12) and the increase in the production of reactive oxygen species may be consequences of the δ-ALA-D inhibition by organochalcogens.
 | ||
| Fig. 12 The consequences of δ-ALA-D inhibition by diphenyl diselenide. The increase of ALA levels can produce reactive oxygen species and impairments in heme biosynthesis, which could disrupt aerobic metabolism. | ||
There is evidence suggesting that modifications of the organic moiety of organochalcogen compounds can have profound effects on their reactivity towards thiols. Accordingly, it has been demonstrated that diorganyl diselenide derived from cholesterol, dicholesteroyl diselenide, does not significantly inhibit the δ-ALA-D activity from different tissues after both in vitro or in vivo exposure.220–222
One of the first demonstrations that the effects of organochalcogens on the δ-ALA-D activity varied depending on the experimental conditions came from a study by Maciel and collaborators.223 In that study, diphenyl diselenide inhibited the δ-ALA-D activity from brain, liver and kidney at a similar potency in vitro. However, when diphenyl diselenide was acutely or chronically administered to mice, the cerebral and hepatic δ-ALA-D activities were inhibited, but the renal enzyme was not altered.223
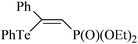
Diethyl 2-phenyl-2-tellurophenyl vinylphosphonate consistently inhibited the cerebral, renal and hepatic δ-ALA-D of mice in vitro. By contrast, exposure of mice for 12 days to this organotellurium compound did not affect the δ-ALA-D from kidney, brain and liver.224 The results indicated that the δ-ALA-D was not a molecular target for diethyl 2-phenyl-2-tellurophenyl vinylphosphonate in vivo, which can be explained by its weak ability to oxidize thiols.
As pointed out above, δ-ALA-D can be a marker of organochalcogen toxicity.203–206 Consequently, the lack of inhibition of the enzyme by organochalcogens may indicate low toxicity of certain compounds. In this context, we have persuasive experimental results demonstrating that organochalcogens that do not inhibit the δ-ALA-D exhibit low toxicity in vivo (Table 3).
| Compound | Animal species | Exposure | Signal of toxicity | Tissue | δ-ALA-D activity | Ref. |
|---|---|---|---|---|---|---|
| a For details of the experimental exposure readers should refer to the references cited above. | ||||||
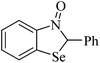
|
Rat | 8 weeks | None | Testes | Normal | 225 |
|
Rat | 8 weeks | None | Testes | Normal | 225 |
|
Rabbit | 8 months | None | Blood, hepatic, cerebral cortex | Increased | 226, 227 |
|
Rabbit | 8 months | None | Renal, hippocampus | Normal | 226, 227 |
|
Mouse | 12 days | None | Renal, hepatic, cerebral | Normal | 224 |
|
Mouse | 72 hours | None | Liver, kidney, brain | Normal | 228 |
|
Rat | 72 hours | None | Hepatic | Normal | 229 |
|
Mouse | 1 hour | None | Brain | Normal | 230 |
|
Rat | 3 days | None | Renal, hepatic, cerebral | Normal | 231 |
Moreover, organochalcogens are effective in reversing hepatic,232–236 cerebral,237 renal238 and pulmonary239 δ-ALA-D activity in experimental models in which this enzyme is inhibited by different xenobiotics. These results reinforcing the hypothesis that δ-ALA-D is not a target of organochalcogens at pharmacological doses.
| Compound | Exposure | Target tissue(s) | ALA-D activity | Signal of toxicity | Ref. |
|---|---|---|---|---|---|
| a For details of the experimental exposure readers should refer to the references cited above. AST – aspartate aminotransferase, ALT – alanine aminotransferase, TG – triglyceride, CAT – catalase, SOD – superoxide dismutase. | |||||
|
250 μmol kg−1, 14 days | Liver | Inhibited | Anemia in mice | 223, 241 |
| PhSeMe | 500 μmol kg−1, 30 days | Liver | Inhibited | Anemia in mice | 242 |
|
300 μmol kg−1, 14 days | Kidney, liver | Inhibited | ↓Body weight gain, ↑AST, ALT in rats | 243 |
|
380 μmol kg−1, one dose | Liver, spleen | Inhibited | ↓Body weight gain, ↓food and water intake in mice | 228 |
|
300 μmol kg−1, one dose | Liver, spleen, kidney, brain | Inhibited | ↓Body weight gain, ↓food and water intake in mice | 228 |
|
75 μmol kg−1, one dose | Liver, spleen | Inhibited | ↓Body weight gain,↑AST, ALT, urea and TG in rats | 244 |
Diphenyl diselenide and its analogues, chloro and trifluoromethyl substituted diaryl diselenides, administered to rodents at doses near to the half maximal lethal dose (LD50) inhibited δ-ALA-D from peripheral tissues and caused signs of systemic and hepatic toxicity (Table 4). Although 1-butyltellurenyl-2-methyl-thioheptene was administered at a single small dose to rats (compared to those used for Se compounds), the animals exhibited signs of systemic toxicity as demonstrated by the reduction of body weight gain, hepatotoxicity (increase of aspartate aminotransferase, AST, and alanine aminotransferase, ALT, activities), renotoxicity (increased urea levels) and dislipidemia (increased serum triglyceride levels) concurrent with the inhibition of δ-ALA-D activity.244 Accordingly, organotellurium compounds have been reported to be more toxic agents to rodents than organoselenium compounds.203,240
As summarized in Table 5 rats exposed to a single high dose of ebselen or diphenyl ditelluride presented signs of renal (increased serum urea) and hepatic toxicity (increased serum AST and ALT) which were coincident with the inhibition of δ-ALA-D from erythrocytes. These observations further suggest that δ-ALA-D can be an enzymatic marker for organochalcogen toxicity.
The potential inhibition of δ-ALA-D activity may lead to neurotoxic effects. Evidence has been found showing that δ-ALA accumulation induces convulsions and that δ-ALA irreversibly inhibits glutamate uptake and is an antagonist of γ-aminobutyric acid (GABA) receptors.245,246 As seen in Table 6, cerebral δ-ALA-D is inhibited by diphenyl diselenide at a dose that caused seizures in rat pups. On the other hand, diphenyl diselenide did not alter cerebral δ-ALA-D activity at anticonvulsant doses.247–249 Accordingly, organoselenium compounds have been reported to have dual effects based on their dose-dependent contrasting action. At low doses, organoselenium has beneficial effects, whereas high doses are toxic. The threshold dose separating these opposing effects has not yet been precisely established.203–206
| Compound | Exposure | Target tissue(s) | δ-ALA-D activity | Signal of toxicity | Ref. |
|---|---|---|---|---|---|
| a For details of the experimental exposure readers should refer to the references cited above. CAT – catalase, SOD – superoxide dismutase. | |||||
|
50 mg kg−1, one dose | Brain, liver | Inhibited | Seizures | 249 |
|
0.03 mg kg−1, 14 days | Hippocampus, striatum | Inhibited | ↑Lipid peroxidation ↓ CAT, SOD | 250 |
The consequences of lactational exposure to diphenyl ditelluride in the brains of suckling rats are shown in Table 6. Diphenyl ditelluride increased lipid peroxidation and inhibited catalase, superoxide dismutase and δ-ALA-D activities in cerebral structures of suckling rats.250 As a consequence of δ-ALA-D inhibition, δ-ALA may undergo autooxidation thus facilitating the generation of reactive oxygen species. The ALA enoyl radical and 4,5-dioxovaleric acid (DOVA), the end products of δ-ALA oxidation, are reactive species that can disrupt cellular prooxidant/antioxidant balance (Fig. 12).68–70 These in vivo observations generate a body of evidence reinforcing that δ-ALA-D from different sources can be an enzymatic marker of toxicity by organochalcogens.
4. δ-ALA-D as a marker of pro-oxidant situations
4.1. δ-ALA-D in animal models of human diseases
Reliable animal models of human diseases are essential for predicting applications in clinical research. In this regard, δ-ALA-D has been indicated as a useful screening marker for pathologies in the humans, mainly because δ-ALA-D activity can be often correlated with oxidative damage on proteins. Thus, it is essential to test its activity in classical animal models of human diseases to clarify whether it translates to real human diseases. Some classical models of diabetes type 1, such as alloxan- and streptozotozin (STZ)-induced diabetes, have already been characterized for δ-ALA-D activity. The results obtained with these drugs were similar to the findings observed in human type 1 diabetes patients in which an inhibition of δ-ALA-D was detected.251–255 Investigating the molecular causes of δ-ALA-D inhibition, it was concluded that it could be caused by the glycation of the active lysine site or by the oxidation of cysteinyl residues of δ-ALA-D.254 Indeed, it has been shown by the reactivation index (with DTT) that this enzyme is more oxidized in diabetic patients.254 Accordingly, a recent work demonstrated that the administration of the antioxidant N-acetylcysteine to diabetic rats restored δ-ALA-D activity to control levels.256Besides diabetes, δ-ALA-D activity has already been linked with a wide variety of other disorders and tested in different animal models of pathologies, such as obesity,257 hypothyroidism,258 hyperglycemia,259 cancer,260 paracetamol intoxication,261 fatigue,262,263 sepsis264 among others.
Obesity and hyperglycemia are of great concern in our society, because they increase susceptibility to development of insulin resistance, diabetes, hypertension and additional complications.265 Animal models of obesity and overweight, usually obtained by high fat diet feeding, have contributed to clarifying many issues related to these complications in the human body. Diet-induced hyperglycemia and high fat diet in animal models have been associated with increased ROS production and with reduced δ-ALA-D activity.257,259 In fact, Folmer et al.266 have demonstrated a strong negative correlation between oxidative stress and δ-ALA-D activity in mice indicating that δ-ALA-D can be an earlier marker of metabolic changes associated with obesity and related pathologies.
Thyroid dysfunction in animal models (induced by propylthiouracil administration) was associated with inhibition of δ-ALA-D.258 Moreover, hypothyroidism may be associated with oxidative stress.267 However, δ-ALA-D activity was found to be higher in patients with hypothyroidism than in patients with normal thyroid function.255 The discrepancies can be related to the time course of disease development (i.e., after acute induction in animal models vs. spontaneous and delayed development in humans), which could allow an adaptation to the metabolic alterations found in humans.
Laboratory animal models of human disorders have been quite helpful in clarifying the putative effects of oxidative stress and other metabolic changes on human δ-ALA-D; however, the number of studies is still limited, indicating that it would be crucial to test the δ-ALA-D activity in other models of human pathologies associated with oxidative stress to determine whether or not δ-ALA-D could be considered a universal marker of oxidative stress.
4.2. δ-ALA-D in human diseases and aging
δ-ALA-D activity is a potential biomarker for screenings of pathophysiological conditions, particularly in those associated with oxidative stress. Accordingly, blood δ-ALA-D has been reported to be inhibited in different pathological conditions, for instance, in hemodialysis patients,268–271 after bone marrow transplantation,272–274 in patients with cervical cancer275 and in diabetes type 1 and 2.251,254,255 Importantly, in various studies, a negative correlation was found between δ-ALA-D activity and oxidative stress. Human δ-ALA-D can also be inhibited after exposure to other pro-oxidant situations, for instance, after hyperoxygenation276 and after exposure to a mixture of solvents in painters.277 This is because δ-ALA-D contains vicinal -SH groups which can be easily oxidized23–26 (Fig. 3 and 4), therefore, inhibiting its activity.278Protein oxidation is a natural phenomenon that occurs in physiological and pathological processes, such as aging and age-associated conditions.279 Aging can be defined either as a normal process of differentiation or as a progressive decrease in physiological function.279 The free radical theory of aging280 has gained experimental support with studies showing an exponential increase in oxidized proteins, lipids and DNA as a function of aging.279 As aging and oxidative damage occurs, δ-ALA-D could be targeted by oxidative stress and be less active in the elderly. For instance, a study comparing young (8 weeks) and aged (38 weeks) mice demonstrated that in aged animals the δ-ALA-D activity was lower than in 8 week-old mice as a consequence of enzyme oxidation with aging.257 Furthermore, δ-ALA-D activity has been suggested as a biomarker of oxidative stress in the elderly population.281 In fact, δ-ALA-D activity decreased as a function of age and there was a negative correlation between age and blood δ-ALA-D activity between 60 to 84 years of age in elderly subjects.281
Age-associated pathologies have been demonstrated to contain altered forms of a variety of proteins, which could be related to protein oxidation and to a decrease in proteases involved in the degradation of aged proteins.279 Protein oxidation is a common observation in age-related pathologies such as Parkinson's282,283 and Alzheimer's diseases.284–286 Interestingly, Alzheimer plaques contain amyloid-β protein, which binds to heme, potentially leading to heme deficiency.287 Moreover, expression of aminolevulinate synthase (ALA-S) and porphobilinogen deaminase (PBG-D) mRNAs have been shown to be reduced in Alzheimer's disease, indicating that different points of heme biosynthesis can be altered in these chronic degenerative pathologies. Unfortunately, δ-ALA-D activity has not yet been tested in Alzheimer's or Parkinson's diseases. However, this enzyme could give important insights into the oxidative state of these age-associated conditions.281
5. Conclusions
δ-ALA-D has been considered a reliable and sensitive marker of Pb(II) exposure. However, in view of the presence of 3 vicinal thiol groups in its active centre, δ-ALA-D can also be oxidized by different soft electrophiles and by metals that compete with Zn(II) at its active centre. Recently, it has been postulated that δ-ALA-D could also be a molecular sensor of oxidative stress. In fact, the enzyme has been reported to be inhibited in experimental and in pathological situations associated with oxidative stress. Remarkably, the substrate of δ-ALA-D, 5-aminolevulinic acid (δ-ALA), is a pro-oxidant compound and δ-ALA-D inhibition can therefore indirectly increase oxidative stress via an increase in the levels of δ-ALA. The oxidative stress triggered by δ-ALA can further inhibit δ-ALA-D, thus setting forth a vicious toxic cycle. In short, δ-ALA-D is more than a biomarker of Pb(II). In fact, it can be considered a biomarker of exposure to different exogenous electrophiles and also to pro-oxidant situations found in relevant human pathologies. Thus, the pair δ-ALA and δ-ALA-D can be considered more than substrate and enzyme, because the inhibition of δ-ALA-D can increase δ-ALA levels that will further inhibit δ-ALA-D, contributing in this way to worsening the redox state of living cells.References
- E. K. Jaffe and S. H. Lawrence, Arch. Biochem. Biophys., 2012, 519, 131–143 CrossRef.
- E. K. Jaffe, Bioorg. Chem., 2004, 32, 316–325 CrossRef CAS.
- S. H. Lawrence, T. Selwood and E. K. Jaffe, ChemMedChem, 2011, 6, 1067–1073 CrossRef CAS.
- G. Layer, J. Reichelt, D. Jahn and D. W. Heinz, Protein Sci., 2010, 19, 1137–1161 CrossRef CAS.
- E. K. Jaffe, J. Bioenerg. Biomembr., 1995, 27, 169–179 CrossRef CAS.
- P. M. Shoolingin-Jordan, P. Spencer, M. Sarwar, P. E. Erskine, K. M. Cheung, J. B. Cooper and E. B. Norton, Biochem. Soc. Trans., 2002, 30, 584–590 CrossRef CAS.
- E. K. Jaffe, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2000, 56, 115–128 CrossRef CAS.
- P. M. Shoolingin-Jordan, Biochem. Soc. Trans., 1998, 26, 326–336 CAS.
- M. J. Warren, J. B. Cooper, S. P. Wood and P. M. Shoolingin-Jordan, Trends Biochem. Sci., 1998, 23, 217–221 CrossRef CAS.
- I. U. Heinemann, M. Jahn and D. Jahn, Arch. Biochem. Biophys., 2008, 474, 238–251 CrossRef CAS.
- S. Sassa, S. Granick and A. Kappas, Ann. N. Y. Acad. Sci., 1975, 244, 419–440 CrossRef CAS.
- H. Fujita, C. Nishitani and K. Ogawa, Tohoku J. Exp. Med., 2002, 196, 53–64 CrossRef CAS.
- S. Sassa, Semin. Liver Dis., 1998, 18, 95–101 CrossRef CAS.
- F. Bardag-Gorce and S. W. French, Exp. Mol. Pathol., 2011, 91, 485–489 CrossRef CAS.
- J. D. Etlinger, S. X. Li, G. G. Guo and N. Li, Enz. Prot., 1993, 47, 325–329 CAS.
- G. G. Guo, M. Gu and J. D. Etlinger, J. Biol. Chem., 1994, 269, 12399–12402 Search PubMed.
- N. Grunberg-Etkovitz, N. Lev, D. Ickowicz, A. Avital, D. Offenand and Z. Malik, J. Environ. Pathol. Toxicol. Oncol., 2009, 28, 5–24 CrossRef CAS.
- N. Grünberg-Etkovitz, L. Greenbaum, B. Grinblat and Z. Malik, Biochim. Biophys. Acta, Mol. Basis Dis., 2006, 1762, 819–827 CrossRef.
- W. H. Wu, D. Shemin, K. E. Richards and R. C. Williams, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 1767–1770 CrossRef CAS.
- E. K. Jaffe, J. Martins, J. Li, J. Kervinen and R. L. Dunbrack Jr., J. Biol. Chem., 2001, 276, 1531–1537 CrossRef CAS.
- P. T. Erskine, E. M. Duke, I. J. Tickle, N. M. Senior, M. J. Warren and J. B. Cooper, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2000, 56, 421–430 CrossRef CAS.
- A. J. Dent, D. Beyersmann, C. Block and S. S. Hasnain, Biochemistry, 1990, 29, 7822–7828 CrossRef CAS.
- G. D. Markham, C. B. Myers, K. A. Harris Jr, M. Volin and E. K. Jaffe, Protein Sci., 1993, 2, 71–79 CrossRef CAS.
- M. Farina, N. B. V. Barbosa, C. W. Nogueira, V. Folmer, G. Zeni, L. H. Andrade, A. L. Braga and J. B. T. Rocha, Braz. J. Med. Biol. Res., 2002, 35, 623–631 CrossRef CAS.
- R. A. Saraiva, D. C. Bueno, P. A. Nogara and J. B. T. Rocha, J. Toxicol. Environ. Health Part A, 2012, 75, 1012–1022 Search PubMed.
- T. Emanuelli, J. B. Rocha, M. E. Pereira, P. C. Nascimento, D. O. Souza and F. A. Beber, Pharmacol. Toxicol., 1998, 83, 95–103 CAS.
- F. A. Beber, J. Wollmeister, M. J. Brigo, M. C. Silva, C. N. Pereira and J. B. Rocha, Int. J. Vitam. Nutr. Res., 1998, 68, 181–188 CAS.
- J. S. Seehra and P. M. Jordan, Eur. J. Biochem., 1981, 113, 435–446 CrossRef CAS.
- J. S. Seehra, M. G. Gore, A. G. Chaudhry and P. M. Jordan, Eur. J. Biochem., 1981, 114, 263–269 CrossRef CAS.
- P. N. Gibbs, M. G. Gore and P. M. Jordan, Biochem. J., 1985, 225, 573–580 CAS.
- P. N. Gibbs, A. G. Chaudhry and P. M. Jordan, Biochem. J., 1985, 230, 25–34 CAS.
- J. B. T. Rocha, A. J. Freitas, M. B. Marques, M. E. Pereira, T. Emanuelli and D. O. Souza, Braz. J. Med. Biol. Res., 1993, 26, 1077–1083 CAS.
- J. R. M. Alves Costa, M. Mela, H. C. da Silva de Assis, E. Pelletier, M. A. F. Randiand and C. A. de Oliveira Ribeiro, Ecotoxicol. Environ. Saf., 2007, 67, 82–88 CrossRef CAS.
- J. B. T. Rocha, S. M. Tuerlinckx, M. R. Schetinger and V. Folmer, Toxicol. Appl. Pharmacol., 2004, 200, 169–176 CrossRef CAS.
- J. B. Rocha, M. E. Pereira, T. Emanuelli, R. S. Christofari and D. O. Souza, Toxicology, 1995, 100, 27–37 CrossRef CAS.
- N. C. Peixoto, T. Roza and M. E. Pereira, Toxicol. in Vitro, 2004, 18, 805–809 CrossRef CAS.
- N. C. Peixoto, T. Roza, E. M. Flores and M. E. Pereira, Toxicol. Lett., 2003, 146, 17–25 CrossRef CAS.
- C. Franciscato, L. Moraes-Silva, F. A. Duarte, C. S. Oliveira, R. P. Ineu, E. M. Flores, V. L. Dressler, N. C. Peixoto and M. E. Pereira, Ecotoxicol. Environ. Saf., 2011, 74, 480–486 CrossRef CAS.
- P. L. Goering and S. Rehm, Environ. Res., 1990, 53, 135–151 CrossRef CAS.
- P. L. Goering, R. R. Maronpotand and B. A. Fowler, Toxicol. Appl. Pharmacol., 1988, 92, 179–193 CrossRef CAS.
- E. A. Conner, H. Yamauchi and B. A. Fowler, Chem.–Biol. Interact., 1995, 96, 273–285 CrossRef CAS.
- P. L. Goering and B. A. Fowler, Arch. Biochem. Biophys., 1987, 253, 48–55 CrossRef CAS , 15.
- P. L. Goering, P. Mistry and B. A. Fowler, J. Pharmacol. Exp. Ther., 1986, 237, 220–225 CAS.
- T. Sakai, S. Yanagihara, Y. Kunugi and K. Ushio, Br. J. Ind. Med., 1983, 40, 61–66 CAS.
- N. L. Pauza, M. J. Cotti, L. Godar, A. M. Ferramola de Sancovich and H. A. Sancovich, J. Inorg. Biochem., 2005, 99, 409–414 CrossRef CAS.
- R. R. Lauwerys, J. P. Buchet and H. A. Roels, Br. J. Ind. Med., 1973, 30, 359–364 CAS.
- S. Tola, S. Hernberg, S. Asp and J. Nikkanen, Br. J. Ind. Med., 1973, 30, 134–141 CAS.
- R. A. Mitchell, J. E. Drake, L. A. Wittlin and T. A. Rejent, Clin. Chem., 1977, 23, 105–111 CAS.
- V. Wagner, M. Wagnerová, D. Wokounová, J. Kriz, Z. Mádlo and O. Mohyla, J. Hyg. Epidemiol. Microbiol. Immunol., 1981, 25, 97–102 CAS.
- H. C. Lichtman and F. Feldman, J. Clin. Invest., 1963, 42, 830–839 CrossRef CAS.
- H. Roels, P. Bruaux, J. P. Buchet, F. Claeys-Thoreau, R. Lauwerys, A. Lafontaine, G. Hubermont and J. Van Overschelde, Arch. Environ. Health, 1976, 31, 310–316 CAS.
- J. Thompson, D. D. Jones and W. H. Beasley, Br. J. Ind. Med., 1977, 34, 32–36 CAS.
- D. Tousoulis, A. Briasoulis, N. Papageorgiou, C. Tsioufis, E. Tsiamis, K. Toutouzas and C. Stefanadis, Recent Pat. Cardiovasc. Drug Discovery, 2011, 6, 103–114 CrossRef CAS.
- M. Valko, D. Leibfritz, J. Moncol, M. T. Cronin, M. Mazur and J. Telser, Int. J. Biochem. Cell Biol., 2007, 39, 44–84 CrossRef CAS.
- K. Jomova, D. Vondrakova, M. Lawson and M. Valko, Mol. Cell. Biochem., 2010, 345, 91–104 CrossRef CAS.
- J. Onuki, M. H. Medeiros, E. J. Bechara and P. Di Mascio, Biochim. Biophys. Acta, Mol. Basis Dis., 1994, 1225, 259–263 CrossRef CAS.
- C. G. Fraga, J. Onuki, F. Lucesoli, E. J. Bechara and P. Di Mascio, Carcinogenesis, 1994, 15, 2241–2244 CrossRef CAS.
- P. I. Oteiza, C. G. Kleinman, M. Demasi and E. J. Bechara, Arch. Biochem. Biophys., 1995, 316, 607–611 CrossRef CAS.
- M. Demasi, C. A. Penatti, R. De Lucia and E. J. Bechara, Free Radical Biol. Med., 1996, 20, 291–299 CrossRef CAS.
- M. Demasi, C. A. Costa, C. Pascual, S. Llesuy and E. J. Bechara, Free Radical Res., 1997, 26, 235–243 CrossRef CAS.
- C. A. Costa, G. C. Trivelato, A. M. Pinto and E. J. Bechara, Clin. Chem., 1997, 43, 1196–1202 CAS.
- M. E. Rocha, F. Dutra, B. Bandy, R. L. Baldini, S. L. Gomes, A. Faljoni-Alário, C. W. Liria, M. T. Miranda and E. J. Bechara, Arch. Biochem. Biophys., 2003, 409, 349–356 CrossRef CAS.
- M. E. Rocha, B. Bandy, C. A. Costa, M. P. de Barros, A. M. Pinto and E. J. Bechara, Free Radical Res., 2000, 32, 343–353 CrossRef CAS.
- C. A. Penatti, E. J. Bechara and M. Demasi, Arch. Biochem. Biophys., 1996, 335, 53–60 CrossRef CAS.
- A. E. Vercesi, R. F. Castilho, A. R. Meinicke, V. G. Valle, M. Hermes-Lima and E. J. Bechara, Biochim. Biophys. Acta, Bioenerg., 1994, 1188, 86–92 CrossRef CAS.
- H. P. Monteiro, D. S. Abdalla, O. Augusto and E. J. Bechara, Arch. Biochem. Biophys., 1989, 271, 206–216 CrossRef CAS.
- B. Pereira, R. Curi, E. Kokubun and E. J. Bechara, J. Appl. Physiol., 1992, 72, 226–230 CAS.
- H. P. Monteiro, E. J. Bechara and D. S. Abdalla, Mol. Cell. Biochem., 1991, 103, 73–83 CrossRef CAS.
- E. J. Bechara, Braz. J. Med. Biol. Res., 1996, 29, 841–851 CAS.
- E. J. Bechara, F. Dutra, V. E. Cardoso, A. Sartori, K. P. Olympio, C. A. Penatti, A. Adhikariand and N. A. Assunção, Comp. Biochem. Physiol., Part C: Toxicol. Pharmacol., 2007, 146, 88–110 CrossRef.
- F. G. Princ, A. G. Maxit, C. Cardalda, A. Batlle and A. A. Juknat, J. Pineal Res., 1998, 24, 1–8 CrossRef CAS.
- E. A. Border, A. C. Cantrell and T. A. Kilroe-Smith, Br. J. Ind. Med., 1976, 33, 85–87 CAS.
- B. Haeger-Aronsen and A. Schütz, Arch. Environ. Health, 1976, 31, 215–220 CAS.
- M. Chiba and M. Kikuchi, Toxicol. Appl. Pharmacol., 1984, 73, 388–394 CrossRef CAS.
- J. R. Davis and M. J. Avram, Toxicol. Appl. Pharmacol., 1978, 44, 181–190 CrossRef CAS.
- M. H. Whittaker, G. Wang, X. Q. Chen, M. Lipsky, D. Smith, R. Gwiazda and B. A. Fowler, Toxicol. Appl. Pharmacol., 2011, 254, 154–166 CrossRef CAS.
- P. Grover, P. V. Rekhadevi, K. Danadevi, S. B. Vuyyuri, M. Mahboob and M. F. Rahman, Int. J. Hyg. Environ. Health, 2010, 213, 99–106 CrossRef CAS.
- Q. Wang, L. X. Ye, H. H. Zhao, J. W. Chen and Y. K. Zhou, Sci. Total Environ., 2011, 409, 1806–1810 CrossRef CAS.
- V. Kasuba, R. Rozgaj, M. Milić, D. Zeljezić, N. Kopjar, A. Pizent and Z. Kljaković-Gaspić, J. Appl. Toxicol., 2010, 30, 321–328 CAS.
- M. Ahamed, S. Verma, A. Kumar and M. K. Siddiqui, Hum. Exp. Toxicol., 2006, 25, 547–553 CAS.
- H. Gurer-Orhan, H. U. Sabir and H. Ozgüneş, Toxicology, 2004, 195, 147–154 CrossRef CAS.
- A. J. Patil, V. R. Bhagwat, J. A. Patil, N. N. Dongre, J. G. Ambekar and K. K. Das, J. Basic Clin. Physiol. Pharmacol., 2006, 17, 213–229 CrossRef CAS.
- I. K. Mohammad, A. A. Mahdi, A. Raviraja, I. Najmul, A. Iqbal and V. Thuppil, Arh. Hig. Rada Toksikol., 2008, 59, 161–169 CrossRef CAS.
- I. Ergurhan-Ilhan, B. Cadir, M. Koyuncu-Arslan, C. Arslan, F. M. Gultepe and G. Ozkan, Pediatr. Int., 2008, 50, 45–50 CrossRef CAS.
- A. Rendón-Ramirez, J. Cerbón-Solórzano, M. Maldonado-Vega, M. A. Quintanar-Escorza and J. V. Calderón-Salinas, Toxicol. in Vitro, 2007, 21, 1121–1126 CrossRef.
- M. Ahamed, M. J. Akhtar, S. Verma, A. Kumar and M. K. Siddiqui, BioScience Trends, 2011, 5, 38–43 CrossRef CAS.
- M. Ahamed, M. Fareed, A. Kumar, W. A. Siddiqui and M. K. J. Siddiqui, Redox Rep., 2008, 13, 117–122 CrossRef CAS.
- G. Zampella, K. P. Neupane, L. De Gioia and V. L. Pecoraro, Chem.–Eur. J., 2012, 18, 2040–2050 CrossRef CAS.
- A. Rosin, Isr. Med. Assoc. J., 2009, 11, 689–694 Search PubMed.
- A. P. Neal and T. R. Guilarte, Mol. Neurobiol., 2010, 42, 151–160 CrossRef CAS.
- M. Jakubowski, Int. J. Occup. Med. Environ. Health, 2011, 24, 1–7 CrossRef.
- P. L. Goering, Neurotoxicology, 1993, 14, 45–60 CAS.
- H. A. Godwin, Curr. Opin. Chem. Biol., 2001, 5, 223–227 CrossRef CAS.
- H. C. Gonick, J. Toxicol., 2011, 2011, 686050 Search PubMed.
- H. Fujita and N. Ishihara, Br. J. Ind, Med., 1988, 45, 710–712 CAS.
- H. Fujita, Y. Orii and S. Sano, Biochim. Biophys. Acta, Gen. Subj., 1981, 678, 39–50 CrossRef CAS.
- H. Fujita, R. Yamamoto, K. Sato and M. Ikeda, Toxicol. Appl. Pharmacol., 1985, 77, 66–75 CrossRef CAS.
- E. C. Goulart, C. A. Pereira, R. C. Garcia, M. B. Giacomelli and A. L. Rodrigues, Braz. J. Med. Biol. Res., 2001, 34, 785–790 CrossRef CAS.
- C. Li, M. Xu, S. Wang, X. Yang, S. Zhou, J. Zhang, Q. Liu and Y. Sun, Toxicol. Lett., 2011, 203, 48–53 CrossRef CAS.
- Y. Nzengue, S. M. Candéias, S. Sauvaigo, T. Douki, A. Favier, W. Rachidi and P. Guiraud, J. Trace Elem. Med. Biol., 2011, 25, 171–180 CAS.
- V. Matović, A. Buha, Z. Bulat and D. Dukić-Ćosić, Arh. Hig. Rada Toksikol., 2011, 62, 65–76 CrossRef.
- R. Khlifi and A. Hamza-Chaffai, Toxicol. Appl. Pharmacol., 2010, 248, 71–88 CrossRef CAS , 15.
- A. Hartwig, BioMetals, 2010, 23, 951–960 CrossRef CAS.
- J. M. Moulis, BioMetals, 2010, 23, 877–896 CrossRef CAS.
- M. M. Braga, T. Dick, D. L. de Oliveira, A. S. Guerra, M. C. Leite, A. P. Ardais, D. O. Souza and J. B. T. Rocha, J. Appl. Toxicol., 2012, 32, 20–25 CrossRef CAS.
- A. P. Ardais, F. W. Santos and C. W. Nogueira, J. Appl. Toxicol., 2008, 28, 322–328 CrossRef CAS.
- R. Sommer and D. Beyersmann, J. Inorg. Biochem., 1984, 20, 131–145 CrossRef CAS.
- M. Schlösser and D. Beyersmann, Biol. Chem. Hoppe-Seyler, 1987, 368, 1469–1477 CrossRef.
- E. K. Jaffe, S. P. Salowe, N. T. Chen and P. A. DeHaven, J. Biol. Chem., 1984, 259, 5032–5036 CAS.
- C. W. Nogueira, F. A. Soares, P. C. Nascimento, D. Muller and J. B. T. Rocha, Toxicology, 2003, 184, 85–95 CrossRef CAS.
- H. A. Roels, J. P. Buchet, R. R. Lauwerys and J. Sonnet, Br. J. Ind. Med., 1975, 32, 181–192 CAS.
- G. M. Conterato, R. P. Bulcão, R. Sobieski, A. M. Moro, M. F. Charão, F. A. Freitas, F. L. Almeida, A. P. Moreira, M. Roehrs, R. Tonello, B. L. Batista, D. Grotto, F. Barbosa Jr, S. C. Garcia and T. Emanuelli, J. Appl. Toxicol., 2011 DOI:10.1002/jat.1731.
- T. D. Seth, L. N. Agarwal, N. K. Satija and M. Z. Hasan, Bull. Environ. Contam. Toxicol., 1976, 16, 190–196 CrossRef CAS.
- P. L. Goering and B. A. Fowler, Biochem. J., 1987, 245, 339–345 CAS.
- R. B. Simpson, J. Am. Chem. Soc., 1961, 83, 4711–4717 CrossRef CAS.
- M. Farina, J. B. T. Rocha and M. Aschner, Life Sci., 2011, 89, 555–563 CrossRef CAS.
- M. Farina, M. Aschner and J. B. T. Rocha, Toxicol. Appl. Pharmacol., 2011, 256, 405–417 CrossRef CAS.
- S. J. B. Fretham, S. Caito , E. J. Martinez-Finley and M. Aschner, Toxicol. Res., 2012, 1, 32–38 RSC.
- A. L. Rodriguez, M. L. Bellinaso and T. Dick, Comp. Biochem. Physiol. B Biochem. Mol. Biol., 1989, 94, 65–69 CrossRef.
- M. Matzapetakis, D. Ghosh, T. C. Weng, J. E. Penner-Hahn and V. L. Pecoraro, JBIC, J. Biol. Inorg. Chem., 2006, 11, 876–890 CrossRef CAS.
- S. J. Yoon, L. M. Diener, P. R. Bloom, E. A. Nater and W. F. Bleam, Geochim. Cosmochim. Acta, 2005, 69, 1111–21 CrossRef CAS.
- S. Bose-O'Reilly, K. M. McCarty, N. Steckling and B. Lettmeier, Curr. Probl. Pediatr. Adolesc. Health Care, 2010, 40, 186–215 CrossRef.
- J. G. Dórea, Int. J. Environ. Res. Public Health, 2010, 7, 3467–3477 CrossRef.
- A. Schutz and S. Skerfving, Scand. J. Work, Environ. Health, 1975, 1, 54–59 CAS.
- D. Grotto, J. Valentini, M. Fillion, C. J. Passos, S. C. Garcia, D. Mergler and F. Barbosa Jr., Sci. Total Environ., 2010, 408, 806–811 CrossRef CAS.
- J. Valentini, J. Vicentini, D. Grotto, R. Tonello, S. C. Garcia and F. Barbosa Jr., Basic Clin. Pharmacol. Toxicol., 2010, 106, 95–99 CrossRef CAS.
- J. S. Woods and B. A. Fowler, J. Lab. Clin. Med., 1977, 90, 266–272 CAS.
- S. D. Pingree, P. L. Simmonds, K. T. Rummel and J. S. Woods, Toxicol. Sci., 2001, 61, 234–240 CrossRef CAS.
- T. Y. Chan, Clin. Toxicol., 2011, 49, 886–891 CrossRef CAS.
- S. Al-Sinani, A. Al-Rawas and A. Dhawan, Clin. Res. Hepatol. Gastroenterol., 2011, 35, 580–582 CrossRef CAS.
- J. J. Berzas Nevado, R. C. Rodríguez Martín-Doimeadios, F. J. Guzmán Bernardo, M. Jiménez Moreno, A. M. Herculano, J. L. do Nascimento and M. E. Crespo-López, Environ. Int., 2010, 36, 593–608 CrossRef CAS.
- M. da Conceição Nascimento, J. L. M. do Nascimento, L. C. de Lima Silveira, J. B. T. Rocha and M. Aschner, Environ. Bioindic., 2009, 4, 222–245 CrossRef.
- C. C. Bridges and R. K. Zalups, J. Toxicol. Environ. Health, Part B, 2010, 13, 385–410 CAS.
- M. Farina, R. Brandão, F. S. de Lara, L. B. Pagliosa, F. A. Soares, D. O. Souza and J. B. T. Rocha, Toxicology, 2003, 184, 179–187 CrossRef CAS.
- T. Emanuelli, J. B. T. Rocha, M. E. Pereira, L. O. Porciuncula, V. M. Morsch, A. F. Martins and D. O. Souza, Pharmacol. Toxicol., 1996, 79, 136–143 CAS.
- J. B. T. Rocha, L. K. Rocha, T. Emanuelli and M. E. Pereira, Toxicol. Lett., 2001, 125, 143–150 CrossRef CAS.
- P. R. Augusti, G. M. M. Conterato, S. Somacal, L. Einsfeld, A. T. Ramos, F. Y. M. Hosomi, D. L. Graça and T. Emanuelli, Basic Clin. Pharmacol. Toxicol., 2007, 100, 398–402 CrossRef CAS.
- P. R. Augusti, G. M. Conterato, S. Somacal, R. Sobieski, P. R. Spohr, J. V. Torres, M. F. Charão, A. M. Moro, M. P. Rocha, S. C. Garcia and T. Emanuelli, Food Chem. Toxicol., 2008, 46, 212–219 CrossRef CAS.
- G. Crisponi, V. M. Nurchi, G. Faa and M. Remelli, Monatsh. Chem., 2011, 142, 331–340 CrossRef CAS.
- M. Kawahara and M. Kato-Negishi, Int. J. Alzheimers Dis., 2011, 2011, 276393 Search PubMed.
- L. Tomljenovic, J. Alzheimers Dis., 2011, 23, 567–598 CAS.
- L. Kaiser and K. A. Schwartz, Am. J. Kidney Dis., 1985, 6, 348–352 CAS.
- T. M. Schroeder and M. L. Caspers, Biochem. Pharmacol., 1996, 52, 927–931 CrossRef CAS.
- M. R. Schetinger, C. D. Bonan, V. M. Morsch, D. Bohrer, L. M. Valentim and S. R. Rodrigues, Braz. J. Med. Biol. Res., 1999, 32, 761–766 CrossRef CAS.
- V. L. Pimentel Vieira, J. B. T. Rocha, M. R. Schetinger, V. M. Morsch, S. R. Rodrigues, S. M. Tuerlinckz, D. Bohrer and P. C. do Nascimento, Toxicol. Lett., 2000, 117, 45–52 CrossRef CAS.
- J. Chmielnicka, M. Nasiadek, E. Lewandowska-Zyndul and R. Pińkowski, Ecotoxicol. Environ. Saf., 1996, 33, 201–206 CrossRef CAS.
- M. Farina, L. N. Rotta, F. A. Soares, F. Jardim, R. Jacques, D. O. Souza and J. B. T. Rocha, Toxicology, 2005, 209, 29–37 CrossRef CAS.
- J. Chmielnicka, M. Nasiadek and E. Lewandowska-Zyndul, Biol. Trace Elem. Res., 1994, 40, 127–136 CrossRef CAS.
- S. J. Flora, A. Mehta, K. Satsangi, G. M. Kannan and M. Gupta, Comp. Biochem. Physiol., Part C: Toxicol. Pharmacol., 2003, 134, 319–328 CrossRef.
- C. R. Chitambar, Int. J. Environ. Res. Public Health, 2010, 7, 2337–2361 CrossRef CAS.
- A. Kiremitci and S. Bolay, J. Oral Rehabil., 2003, 30, 664–667 CrossRef CAS.
- S. J. Flora and S. Das Gupta, J. Appl. Toxicol., 1992, 12, 333–334 CrossRef CAS.
- S. J. Flora, P. Kumar, G. M. Kannan and G. P. Rai, Toxicol. Lett., 1998, 94, 103–113 CrossRef CAS.
- S. J. Flora, S. N. Dube, R. Vijayaraghavan and S. C. Pant, Biol. Trace Elem. Res., 1997, 58, 197–208 CrossRef CAS.
- S. J. Flora, K. Bhatt, N. Dwivedi, V. Pachauri and P. K. Kushwah, Clin. Exp. Pharmacol. Physiol., 2011, 38, 373–379 CrossRef CAS.
- K. Bhatt and S. J. Flora, Environ. Toxicol. Pharmacol., 2009, 28, 140–146 CrossRef CAS.
- S. J. Flora, A. Mehta, P. V. Rao, G. M. Kannan, A. S. Bhaskar, S. N. Dube and B. P. Pant, Toxicology, 2004, 195, 127–146 CrossRef CAS.
- S. J. Flora, R. Dubey, G. M. Kannan, R. S. Chauhan, B. P. Pant and D. K. Jaiswal, Toxicol. Lett., 2002, 132, 9–17 CrossRef CAS.
- S. J. Flora, G. M. Kannan and P. Kumar, Chem.-Biol. Interact., 1999, 122, 1–13 CrossRef CAS.
- A. Tanaka, Toxicol. Appl. Pharmacol., 2004, 198, 405–411 CrossRef CAS.
- P. Cvjetko, I. Cvjetko and M. Pavlica, Arh. Hig. Rada Toksikol., 2010, 61, 111–119 CrossRef CAS.
- J. S. Woods and B. A. Fowler, Toxicol. Appl. Pharmacol., 1986, 83, 218–229 CrossRef CAS.
- J. Ventura-Lima, M. R. Bogo and J. M. Monserrat, Ecotoxicol. Environ. Saf., 2011, 74, 211–218 CrossRef CAS.
- M. F. Hughes, Toxicol. Lett., 2002, 133, 1–16 CrossRef CAS.
- A. Vahidnia, G. B. van derVoet and F. A. de Wolff, Hum. Exp. Toxicol., 2007, 26, 823–832 CAS.
- M. Modi, R. K. Kaul, G. M. Kannan and S. J. Flora, J. Trace Elem. Med. Biol., 2006, 20, 197–204 CAS.
- M. Modi, U. Pathak, K. Kalia and S. J. Flora, Environ. Toxicol. Pharmacol., 2005, 19, 131–138 CrossRef CAS.
- M. Mittal and S. J. Flora, Chem.-Biol. Interact., 2006, 162, 128–139 CrossRef CAS.
- C. Chandronitha, S. Ananthi, G. Ramakrishnan, R. Lakshmisundaram, V. Gayathri and H. R. Vasanthi, Hum. Exp. Toxicol., 2010, 29, 705–719 CAS.
- S. J. Flora, S. Chouhan, G. M. Kannan, M. Mittal and H. Swarnkar, Oxid. Med. Cell. Longevity, 2008, 1, 39–45 CrossRef.
- G. SharmilaBanu, G. Kumar and A. G. Murugesan, Food Chem. Toxicol., 2009, 47, 490–495 CrossRef CAS.
- D. Srivastava, R. B. Subramanian, D. Madamwarand and S. J. Flora, Arh. Hig. Rada Toksikol., 2010, 61, 153–159 CrossRef CAS.
- D. Mishra and S. J. Flora, Biol. Trace Elem. Res., 2008, 122, 137–147 CrossRef CAS.
- H. K. Okoro, O. S. Fatoki, F. A. Adekola, B. J. Ximba, R. G. Snyman and B. Opeolu, Rev. Environ. Contam. Toxicol., 2011, 213, 27–54 CAS.
- A. Trabucco, P. Di Pietro, S. L. Nori, F. Fulceri, L. Fumagalli, A. Paparelliand and F. Fornai, Arch. Ital. Biol., 2009, 147, 141–153 CAS.
- B. Michalke, S. Halbach and V. Nischwitz, J. Environ. Monit., 2009, 11, 939–954 RSC.
- M. I. Yousef, T. I. Awad, F. A. Elhag and F. A. Khaled, Toxicology, 2007, 235, 194–202 CrossRef CAS.
- J. Chmielnicka, G. Zareba and U. Grabowska, Ecotoxicol. Environ. Saf., 1992, 24, 266–274 CrossRef CAS.
- G. Zareba and J. Chmielnicka, Ecotoxicol. Environ. Saf., 1985, 9, 40–46 CrossRef CAS.
- G. Zareba, J. Chmielnicka and G. Kustra, Ecotoxicol. Environ. Saf., 1986, 11, 144–152 CrossRef CAS.
- M. Chiba and M. Kikuchi, Br. J. Ind. Med., 1979, 36, 323–325 CAS.
- M. Chiba, N. Fujimoto and M. Kikuchi, Toxicol. Lett., 1985, 24, 235–241 CrossRef CAS.
- H. Ovaska, D. M. Wood, I. House, P. I. Dargan, A. L. Jones and S. Murray, Clin. Toxicol., 2008, 46, 855–857 CrossRef CAS.
- Z. Chen, Q. Zhou and R. Ge, BioMetals, 2012, 25, 95–102 CrossRef CAS.
- J. S. Woods and B. A. Fowler, Toxicol. Appl. Pharmacol., 1987, 90, 274–283 CrossRef CAS.
- W. J. Rogan, J. R. Reigart and B. C. Gladen, J. Pediatr., 1986, 109, 60–64 CrossRef CAS.
- M. Bergomi, P. Borella, G. Fantuzzi, G. Vivoli, N. Sturloni, G. Cavazzuti, A. Tampieri and P. L. Tartoni, Dev. Med. Child Neurol., 1989, 31, 181–190 CAS.
- R. de C. Mattos, M. A. Carvalho, H. R. Mainenti, E. C. Xavier Junior, P. de N. Sarcinelli, L. B. Carvalho, R. M. Borges, S. L. Quiterio, S. M. Nogueira, I. C. Costa and M. de F. Alves, Cienc. Saude Coletiva, 2009, 14, 2039–2048 CrossRef.
- O. P. Soldin, B. Hanak and S. J. Soldin, Clin. Chim. Acta, 2003, 327, 109–113 CrossRef CAS.
- M. Ahamed and M. K. Siddiqui, Clin. Nutr., 2007, 26, 400–408 CrossRef CAS.
- L. Lopez-Carrillo, L. Torres-Sanchez, F. Garrido, J. Papaqui-Hernandez, E. Palazuelos-Rendon and M. Lopez-Cervantes, Environ. Health Perspect., 1996, 104, 1208–1211 CAS.
- T. Sakai, Ind. Health, 2000, 38, 127–142 CrossRef CAS.
- N. Pawlas, K. Broberg, E. Olewinska, A. Prokopowicz, S. Skerfving and K. Pawlas, NeuroToxicology, 2011, 33, 37–43 CrossRef.
- J. G. Wetmur, G. Lehnert and R. J. Desnick, Environ. Res., 1991, 56, 109–119 CrossRef CAS.
- J. G. Wetmur, A. H. Kaya, M. Plewinska and R. J. Desnick, Am. J. Hum. Genet., 1991, 49, 757–763 CAS.
- A. P. Shaik and K. Jamil, Toxicol. Ind. Health, 2008, 24, 501–506 CrossRef CAS.
- F. Scinicariello, H. E. Murray, D. B. Moffett, H. G. Abadin, M. J. Sexton and B. A. Fowler, Environ. Health Perspect., 2007, 115, 35–41 CrossRef CAS.
- F. Perez-Bravo, M. Ruz, M. J. Moran-Jimenez, M. Olivares, A. Rebolledo, J. Codoceo, V. Sepulveda, A. Jenkin, J. L. Santos and A. Fontanellas, Arch. Environ. Contam. Toxicol., 2004, 47, 276–280 CrossRef CAS.
- Y. Seko, Y. Saito, J. Kitahara and N. Imura, in Selenium in Biology and Medicine, ed. A. Wendel, Springer-Verlag, Berlin, 1989, pp. 70–73 Search PubMed.
- J. Kitahara, Y. Seko and N. Imura, Arch. Toxicol., 1993, 67, 497–501 CrossRef CAS.
- J. E. Spallholz, Free Radical Biol. Med., 1994, 17, 45–64 CrossRef CAS.
- Y. Lin and J. E. Spallholz, Biochem. Pharmacol., 1993, 45, 429–437 CrossRef.
- C. W. Nogueira, G. Zeni and J. B. T. Rocha, Chem. Rev., 2004, 10, 6255–6285 CrossRef.
- C. W. Nogueira and J. B. T. Rocha, J. Braz. Chem. Soc., 2010, 21, 2055–2071 CrossRef CAS.
- C. W. Nogueira and J. B. T. Rocha, Arch. Toxicol., 2011, 85, 1313–1359 CrossRef CAS.
- C. W. Nogueira and J. B. T. Rocha, in PATAI'S Chemistry of Functional Groups; The Chemistry of Organic Selenium and Tellurium Compounds, ed. Z. Rappoport, John Wiley & Sons, DOI:10.1002/9780470682531.pat0567.
- N. B. V. Barbosa, J. B. T. Rocha, G. Zeni, T. Emanuelli, M. C. Beque and A. L. Braga, Toxicol. Appl. Pharmacol., 1998, 149, 243–253 CrossRef CAS.
- C. W. Nogueira, V. C. Borges, G. Zeni and J. B. T. Rocha, Toxicology, 2003, 191, 169–178 CrossRef CAS.
- R. M. Golombieski, D. A. S. Graichen, L. A. Pivetta, C. W. Nogueira, E. L. S. Loreto and J. B. T. Rocha, Comp. Biochem. Physiol., Part C: Toxicol. Pharmacol., 2008, 147, 198–204 CrossRef CAS.
- F. A. Soares, M. Farina, A. C. Boettcher, A. L. Braga and J. B. T. Rocha, Environ. Res., 2005, 98, 46–54 CrossRef.
- E. K. Jaffe, S. Ali, L. W. Mitchell, K. M. Taylor, M. Volin and G. D. Markham, Biochemistry, 1995, 10, 244–251 CrossRef.
- M. Farina, V. Folmer, R. C. Bolzan, L. H. Andrade, G. Zeni, A. L. Braga and J. B. T. Rocha, Toxicol. Lett., 2001, 119, 27–37 CrossRef CAS.
- R. C. Bolzan, V. Folmer, M. Farina, G. Zeni, C. W. Nogueira, J. B. T. Rocha and T. Emanuelli, Pharmacol. Toxicol., 2002, 90, 214–219 CAS.
- D. E. Goeger and H. E. Ganther, Arch. Biochem. Biophys., 1994, 448–451 CrossRef CAS.
- V. Folmer, R. C. Bolzan, M. Farina, G. Zeni, C. W. Nogueira, T. Emanuelli and J. B. T. Rocha, Toxicology, 2005, 206, 403–411 CrossRef CAS.
- H. C. Braga, H. A. Stefani, M. W. Paixao, F. W. Santos and D. S. Ludtke, Tetrahedron, 2010, 66, 3441–3446 CrossRef CAS.
- C. A. Bruning, M. Prigol, D. A. Barancelli, C. W. Nogueira and G. Zeni, Mol. Cell. Biochem., 2009, 332, 17–24 CrossRef.
- A. C. G. Souza, C. Luchese, J. S. S. Neto and C. W. Nogueira, Life Sci., 2009, 84, 351–357 CrossRef CAS.
- V. C. Borges, G. Dadalt, L. Savegnago, A. V. Moro, J. B. T. Rocha and C. W. Nogueira, Toxicol. in Vitro, 2007, 21, 387–391 CrossRef CAS.
- I. J. Kade, M. W. Paixao, O. E. D. Rodrigues, N. B. V. Barbosa, A. L. Braga, D. S. Avila, C. W. Nogueira and J. B. T. Rocha, Neurochem. Res., 2008, 33, 167–178 CrossRef CAS.
- I. J. Kade, M. W. Paixao, O. E. D. Rodrigues, E. O. Ibukun, A. L. Braga, G. Zeni, C. W. Nogueira and J. B. T. Rocha, Toxicol. in Vitro, 2009, 23, 14–20 CrossRef CAS.
- I. J. Kade and J. B. T. Rocha, J. Appl. Toxicol., 2010, 30, 688–693 CrossRef CAS.
- E. N. Maciel, R. C. Bolzan, A. L. Braga and J. B. T. Rocha, J. Biochem. Mol. Toxicol., 2000, 14, 310–319 CrossRef CAS.
- D. S. de Avila, M. C. Beque, V. Folmer, A. L. Braga, G. Zeni, C. W. Nogueira, F. A. A. Soares and J. B. T. Rocha, Toxicology, 2006, 224, 100–107 CrossRef.
- E. C. Stangherlin, A. M. Favero, S. N. Weis, G. Zeni, J. B. T. Rocha and C. W. Nogueira, Food Chem. Toxicol., 2006, 44, 662–669 CrossRef CAS.
- A. F. de Bem, R. D. L. Portella, J. Perottoni, E. Becker, D. Bohrer, M. W. Paixao, C. W. Nogueira, G. Zeni and J. B. T. Rocha, Chem.–Biol. Inter., 2006, 162, 1–10 CrossRef.
- A. F. de Bem, R. D. L. Portella, J. Perottoni, M. W. Paixao, C. W. Nogueira and J. B. T. Rocha, Basic Clin. Pharmacol. Toxicol., 2007, 101, 47–55 CrossRef.
- L. Savegnago, C. R. Jesse and C. W. Nogueira, Environ. Toxicol. Pharmacol., 2009, 27, 271–276 CrossRef CAS.
- V. C. Borges, L. Savegnago, S. Pinton, C. R. Jesse, D. Alves and C. W. Nogueira, J. Appl. Toxicol., 2008, 28, 839–848 CrossRef CAS.
- E. A. Wilhelm, C. R. Jesse, S. S. Roman, C. F. Bortolatto and C. W. Nogueira, Life Sci., 2010, 87, 19–22 CrossRef.
- W. Hassan, S. Pinton, J. T. da Rocha, A. M. Deobald, A. L. Braga, C. W. Nogueira, A. S. Latini and J. B. T. Rocha, Chem.-Biol. Interact., 2011, 190, 35–44 CrossRef CAS.
- E. A. Wilhelm, C. R. Jesse, S. S. Roman, C. W. Nogueira and L. Savegnago, Exp. Mol. Pathol., 2009, 87, 20–26 CrossRef CAS.
- E. A. Wilhelm, C. Jesse, M. Leite and C. W. Nogueira, Pathophysiology, 2009, 16, 31–37 CrossRef CAS.
- M. Ibrahim, M. Prigol, W. Hassan, C. W. Nogueira and J. B. T. Rocha, Cell Biochem. Funct., 2010, 28, 258–265 CrossRef CAS.
- E. A. Wilhelm, C. R. Jesse, C. F. Bortolatto and C. W. Nogueira, Fundam. Clin. Pharmacol., 2011, 25, 80–89 CrossRef CAS.
- E. A. Wilhelm, C. R. Jesse, M. Prigol, D. Alves, R. F. Schumacher and C. W. Nogueira, Cell Biol. Toxicol., 2010, 26, 569–577 CrossRef CAS.
- M. Prigol, C. A. Bruning, G. Zeni and C. W. Nogueira, Biochem. Eng. J., 2009, 45, 94–99 CrossRef CAS.
- R. Brandao, C. I. Acker, M. R. Leite, N. B. V. Barbosa and C. W. Nogueira, J. Appl. Toxicol., 2009, 29, 612–618 CrossRef CAS.
- C. Luchese, E. A. Stangherlin, A. P. Ardais, C. W. Nogueira and F. W. Santos, Toxicology, 2007, 230, 189–196 CrossRef CAS.
- F. C. Meotti, V. C. Borges, G. Zeni, J. B. T. Rocha and C. W. Nogueira, Toxicol. Lett., 2003, 143, 9–16 CrossRef CAS.
- M. C. Jacques-Silva, C. W. Nogueira, L. C. Broch, E. M. M. Flores and J. B. T. Rocha, Pharmacol. Toxicol., 2001, 88, 119–125 CAS.
- V. Folmer, M. Farina, E. N. Maciel, C. W. Nogueira, G. Zeni, T. Emanuelli and J. B. T. Rocha, Drug Chem. Toxicol., 2004, 27, 331–340 CrossRef CAS.
- F. C. Meotti, V. C. Borges, J. Perottoni and C. W. Nogueira, J. Appl. Toxicol., 2008, 28, 638–644 CrossRef CAS.
- L. Savegnago, V. C. Borges, D. Alves, C. R. Jesse, J. B. T. Rocha and C. W. Nogueira, Life Sci., 2006, 79, 1546–1552 CrossRef CAS.
- T. Emanuelli, C. C. P. Prauchner, J. Dacanal, A. Zeni, E. C. Reis, C. F. Mello and D. O. Souza, Brain Res., 2000, 868, 88–94 CrossRef CAS.
- T. Emanuelli, F. W. Pagel, L. O. Porciuncula and D. O. Souza, Neurochem. Int., 2003, 42, 115–121 CrossRef CAS.
- M. Prigol, E. A. Wilhelm, E. C. Stangherlin, D. A. Barancelli, C. W. Nogueira and G. Zeni, Neurochem. Res., 2008, 33, 996–1004 CrossRef CAS.
- M. Prigol, E. A. Wilhelm, C. C. Schneider, J. B. T. Rocha, C. W. Nogueira and G. Zeni, Brain Res., 2007, 1147, 226–232 CrossRef CAS.
- M. Prigol, C. A. Bruning, C. W. Nogueira and G. Zeni, Chem.-Biol. Interact., 2011, 193, 65–70 CrossRef CAS.
- E. C. Stangherlin, A. P. Ardais, J. B. T. Rocha and C. W. Nogueira, Arch. Toxicol., 2009, 83, 485–491 CrossRef CAS.
- B. Fernandez-Cuartero, J. L. Rebollar, A. Batlle and R. Enriquez de Salamanca, Int. J. Biochem. Cell Biol., 1999, 31, 479–488 CrossRef CAS.
- N. B. Barbosa, C. Oliveira, D. Araldi, V. Folmer, J. B. Rocha and C. W. Nogueira, Biol. Pharm. Bull., 2008, 31, 2200–2204 CAS.
- F. Caballero, E. Gerez, A. Batlle and E. Vazquez, Chem.-Biol. Interact., 2000, 126, 215–25 CrossRef CAS.
- G. Bonfanti, R. B. Ceolin, T. Valcorte, K. S. De Bona, L. de Lucca, T. L. Gonçalves and M. B. Moretto, Clin. Biochem., 2011, 44, 1105–1109 CrossRef CAS.
- J. B. Souza, J. B. T. Rocha, C. W. Nogueira, V. C. Borges, R. R. Kaizer, V. M. Morsch, V. L. Dressler, A. F. Martins, E. M. Flores and M. R. Schetinger, Clin. Biochem., 2007, 40, 321–325 CrossRef CAS.
- G. Ribeiro, M. Roehrs, A. Bairros, A. Moro, M. Charao, F. Araujo, J. Valentini, M. Arbo, N. Brucker, R. Moresco, M. Leal, V. Morsch and S. C. Garcia, Drug Chem. Toxicol., 2011, 34, 467–474 CrossRef CAS.
- V. B. Brito, V. Folmer, J. C. Soares, I. D. Silveira and J. B. T. Rocha, Nutrition, 2007, 23, 818–826 CrossRef CAS.
- F. S. Gravina, C. K. da Silveira, A. M. de Assis, D. K. Rieger, C. Guerini, A. P. Muller, M. Farina, L. N. Rotta and M. L. Perry, Exp. Biol. Med., 2007, 232, 1021–1026 CrossRef CAS.
- V. Folmer, J. C. Soares and J. B. T. Rocha, Int. J. Biochem. Cell Biol., 2002, 34, 1279–1285 CrossRef CAS.
- A. Casas, H. Fukuda, P. Riley and A. M. del C. Batlle, Cancer Lett., 1997, 121, 105–113 CrossRef CAS.
- M. T. Olaleye and J. B. Rocha, Exp. Toxicol. Pathol., 2008, 59, 319–327 CrossRef CAS.
- J. C. Soares, V. Folmer and J. B. Rocha, Nutrition, 2003, 19, 627–632 CrossRef CAS.
- T. Tahara, M. Tanaka, S. Nozaki, G. Jin, H. Onoe and Y. Watanabe, Biochem. Biophys. Res. Commun., 2007, 353, 1068–1073 CrossRef CAS.
- C. A. Prauchner, A. S. de Prestes and J. B. Rocha, Pathol., Res. Prac., 2011, 207, 554–558 CrossRef CAS.
- M. T. Zanella, O. Kohlmann Jr. and A. B. Ribeiro, Hypertension, 2001, 38, 705–708 CrossRef CAS.
- V. Folmer, J. C. Soares, D. Gabriel and J. B. T. Rocha, J. Nutr., 2003, 133, 2165–2170 CAS.
- L. Fayadat, P. Niccoli-Sire, J. Lanet and J. L. Franc, J. Biol. Chem., 1999, 274, 10533–10538 CrossRef CAS.
- S. C. Garcia, A. T. Wyse, J. Valentini, M. Roehrs, A. M. Moro, C. Paniz, G. Schmitt, D. Grotto and V. J. Pomblum, Clin. Biochem., 2008, 41, 474–479 CrossRef CAS.
- J. Valentini, D. Grotto, C. Paniz, M. Roehrs, G. Burg and S. C. Garcia, Biomed. Pharmacother., 2008, 62, 378–382 CrossRef CAS.
- M. Roehrs, J. Valentini, R. Bulcao, J. C. Moreira, H. Biesalski, R. P. Limberger, T. Grune and S. C. Garcia, Nephrol., Dial., Transplant., 2009, 24, 2212–2218 CrossRef CAS.
- A. C. da Silva, J. B. Rocha, A. L. Morsch, R. F. Zanin, R. Kaizer, P. A. Maldonado, L. C. Arantes, L. A. Silva, V. M. Morsch and M. R. Schetinger, Biomed. Pharmacother., 2007, 61, 180–185 CrossRef CAS.
- T. L. Gonçalves, D. M. Benvegnú, G. Bonfanti, A. V. Frediani and J. B. Rocha, Pharmacol. Res., 2009, 59, 279–284 CrossRef.
- T. L. Gonçalves, D. M. Benvegnu, G. Bonfanti, A. V. Frediani and J. B. T. Rocha, BMC Cancer, 2009, 9, 138 CrossRef.
- T. L. Gonçalves, D. M. Benvegnú, G. Bonfanti, A. V. Frediani, D. V. Pereira and J. B. Rocha, Clin. Biochem., 2009, 42, 602–610 CrossRef.
- T. L. Gonçalves, F. Erthal, C. L. Corte, L. G. Müller, C. M. Piovezan, C. W. Nogueira and J. B. Rocha, Clin. Biochem., 2005, 38, 1071–1075 CrossRef.
- J. B. Rocha, N. M. H. Bulow, E. F. Correa, C. Scholze, C. W. Nogueira and N. B. Barbosa, Hum. Exp. Toxicol., 2011, 30, 289–295 CAS.
- A. M. Moro, M. Charão, N. Brucker, R. Bulcão, F. Freitas, G. Guerreiro, M. Baierle, S. Nascimento, F. Waechter, V. Hirakata, R. Linden, F. V. Thiesen and S. C. Garcia, Sci. Total Environ., 2010, 408, 4461–4467 CrossRef CAS.
- J. Valentini, G. C. Schmitt, D. Grotto, L. D. Santa Maria, S. P. Boeira, S. J. Piva, N. Brucker, D. Bohrer, V. J. Pomblum, T. Emanuelli and S. C. Garcia, Clin. Biochem., 2007, 40, 591–594 CrossRef CAS.
- E. R. Stadtman, Ann. N. Y. Acad. Sci., 2001, 928, 22–38 CrossRef CAS.
- D. Harman, J. Gerontol., 1956, 11, 298–300 CAS.
- C. Paniz, A. Bairros, J. Valentini, M. Charao, R. Bulcao, A. Moro, T. Grune and S. C. Garcia, Clin. Biochem., 2007, 40, 1367–1372 CrossRef CAS.
- A. Yoritaka, N. Hattori, K. Uchida, M. Tanaka, E. R. Stadtman and Y. Mizuno, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 2696–2701 CrossRef CAS.
- G. Watfa, C. Dragonas, T. Brosche, R. Dittrich, C. C. Sieber, C. Alecu, A. Benetos and R. Nzietchueng, J. Nutr., Health Aging, 2011, 15, 277–281 CrossRef CAS.
- M. E. Harris, K. Hensley, D. A. Butterfield, R. A. Leedle and J. M. Carney, Exp. Neurol., 1995, 131, 193–202 CrossRef CAS.
- M. A. Smith, M. Rudnicka-Nawrot, P. L. Richey, D. Praprotnik, P. Mulvihill, C. A. Miller, L. M. Sayre and G. Perry, J. Neurochem., 1995, 64, 2660–2666 CrossRef CAS.
- J. Greilberger, D. Fuchs, F. Leblhuber, M. Greilberger, R. Wintersteiger and E. Tafeit, Clin. Lab., 2010, 56, 441–448 CAS.
- H. Atamna and W. H. Frey, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11153–11158 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |