Induction of plasma acetylcholinesterase activity and apoptosis in mice treated with the organophosphorus toxicant, tri-o-cresyl phosphate
Wei
Jiang
a,
Ellen G.
Duysen
ab and
Oksana
Lockridge
*b
aDepartment of Environmental, Agricultural and Occupational Health, University of Nebraska Medical Center, Omaha, NE, USA. E-mail: wjiang@unmc.edu; ellen.duysen@unmc.edu
bEppley Institute, University of Nebraska Medical Center, Omaha, NE 68198-5950, USA. E-mail: olockrid@unmc.edu; Fax: +1 402 559 4651; Tel: +1 402 559 6032
First published on 16th April 2012
Abstract
Organophosphorus compounds (OP) inhibit acetylcholinesterase (AChE) activity and cause cultured cells to undergo apoptosis. Live mice treated with OP have reduced AChE activity, but after a short recovery period, their AChE activity rebounds to levels that exceed baseline by more than 2-fold. To date no information is available on whether abnormally high AChE activity is characteristic of apoptosis in animals. Our goal was to determine whether induction of AChE activity is associated with apoptosis in live mice. For this purpose we treated mice with 1500 mg kg−1 tri-o-cresyl phosphate. On day one after treatment their plasma AChE activity was inhibited by 50%. On day 4, plasma AChE activity rebounded to a level 2.2-fold higher than pretreatment activity and remained elevated for about two months. On day 4, AChE activity in the lung was 1.5-fold higher than in controls. Cells in lung sections that were positive in the apoptosis TUNEL assay, stained heavily for AChE activity. In conclusion, AChE activity and apoptosis are induced in mice treated with tri-o-cresyl phosphate. Unusually high AChE activity may be a marker of exposure to apoptosis-inducing substances.
Introduction
Organophosphorus (OP) compounds irreversibly inhibit acetylcholinesterase (AChE) activity resulting in acute toxicity. It is generally assumed that AChE activity will return to normal levels due to synthesis of new enzyme,1 but will not exceed this level. The fact that OP compounds inhibit AChE activity is so firmly established that scientists have not considered the possibility that OP could also induce AChE activity. We demonstrated previously that plasma AChE activity was initially inhibited in living mice treated with non-lethal doses of chlorpyrifos, chlorpyrifos oxon, diazinon, parathion, dichlorvos, tri-o-cresyl phosphate, tri-cresyl phosphate, tabun thiocholine, and diisopropylfluorophosphate, but after 1 to 25 days plasma AChE activity rebounded to levels that exceeded baseline activity by 1.5 to 2.5-fold.2 Plasma butyrylcholinesterase (BChE) activity was also inhibited in OP-treated mice, but plasma BChE activity never rebounded to levels above baseline. In our previous study we did not examine the possibility that induced AChE activity was associated with apoptosis.Rebound of AChE activity to levels above baseline has been reported by other groups. Rats dosed weekly for 4 weeks with chlorpyrifos had reduced AChE activity, but 15% higher levels of AChE protein in the brain compared to control.3 Rats treated intraperitoneally with 100 mg kg−1 malathion for 3 consecutive days had 170% of normal AChE activity in the cerebral cortex and 150% in the hippocampus.4 Administration of low doses of physostigmine and sarin daily for 30 days to rats, resulted in a rise of AChE activity to 143% of normal in plasma on day 61, and a rise of AChE activity to 147% of normal in brain on day 31.5
OP compounds cause apoptosis in cultured cells.6–11 Cultured cells which had no detectable AChE activity under normal culture conditions acquired AChE activity, AChE protein, and AChE mRNA when subjected to conditions that stimulated apoptosis, namely serum starvation or treatment with daunorubicin.12 Our goal was to test the hypothesis that the increased plasma AChE activity in living mice treated with tri-o-cresyl phosphate was associated with apoptosis.
In the current study, we selected tri-o-cresyl phosphate (TOCP) for the organophosphate because TOCP does not cause obvious toxic symptoms in mice. Tri-o-cresyl phosphate is a component of tri-cresyl phosphate, which is a mixture of cresyl phosphate isomers. Tri-cresyl phosphate is used as a plasticizer, a flame retardant, and an anti-wear agent in jet engine oil and hydraulic fluid.13 Studies with rat liver microsomes have shown that TOCP is activated to 2-(o-cresyl)-4H-1,3,2-benzodioxaphosphoran-2-one (also called cresyl saligenin phosphate) in vivo by cytochrome P450. Cresyl saligenin phosphate is known to inhibit cholinesterase and neuropathy target esterase.14,15 Low-level TOCP exposure is hypothesized to be associated with aerotoxic syndrome in jet airline workers, though this association has not been proven.16,17 Humans who ingest high doses of TOCP develop paralysis of the limbs.18
We observed a direct relationship between apoptosis and AChE activity in mouse lung. This is the first report to show that OP exposure induces excess AChE activity and apoptosis in animals.
Materials and methods
Reagents
Tri-o-tolyl phosphate (CAS-78-30-8), also called tri-o-cresyl phosphate, was purchased from Acros Organics, part of Thermo Fisher Scientific (cat# 366460250, Lot A0250944, 96%). The following were purchased from Sigma (St. Louis, MO): ethopropazine hydrochloride (cat# E-2880); tetraisopropyl pyrophosphoramide (cat# T-1505). Lithium heparin microvettes 300 LH were from Sarstedt (Newton, NC). Tissue-Tek O.C.T. (Optimal Cutting Temperature compound cat# 4583) was from Sakura Finetek Inc. (Torrance, CA). The In Situ Apoptosis Detection Kit #MK500 by TAKARA BIO INC. was purchased from Clontech Laboratories Inc. (Madison, WI).Mice
Animal work was conducted in accordance with the Guide for the Care and Use of Laboratory Animals as adopted by the National Institutes of Health. Formal approval to conduct the experiments was obtained from the Institutional Animal Care and Use Committee of the University of Nebraska Medical Center. Mice in strain C57BL/6 were bred at the University of Nebraska Medical Center from parental ES1−/− plasma carboxylesterase knockouts19 and BChE−/− butyrylcholinesterase knockouts20 to produce the ES1−/−BChE+/− genotype. The ES1−/− mice are completely deficient in plasma carboxylesterase activity, but have normal carboxylesterase activity in liver, intestine, and other tissues because the carboxylesterase in tissues is encoded by genes distinct from the plasma carboxylesterase gene that was knocked out.19 Adult ES1−/−BChE+/− mice had approximately one half of wild type BChE activity levels and normal AChE activity. The mice were healthy and fertile.ES1−/− and BChE−/− knockout mice were submitted to The Jackson Laboratory Repository (Bar Harbor, ME) http://jaxmice.jax.org/query. The plasma carboxylesterase knockouts are listed as JAX Stock No. 014096 strain name C57BL/6-Ces1ctm1.1Loc/J. The butyrylcholinesterase knockouts are listed as JAX Stock No. 008087 strain name B6.129S1-Bchetm1Loc/J. The Jackson Laboratory provides heterozygous animals that must be bred to produce completely deficient animals.
Mouse bedding was changed once a week for the first two weeks of the trial with TOCP, and then daily after it was noticed that AChE activity levels in plasma fluctuated. The more frequent change of bedding was intended to minimize the effect of coprophagic behavior, which could potentially re-expose mice to TOCP.
Challenge with tri-o-cresyl phosphate
Adult male ES1−/−BChE+/− mice (n = 4 treatment; n = 4 control) were treated subcutaneously (sc) with a single dose of 1500 mg kg−1 tri-o-cresyl phosphate (TOCP). This dose required 37.5 μl of undiluted TOCP oil per 25 g mouse. Control mice were treated sc with 37.5 μl ethanol. Blood (50 μl) from the treatment and control groups was collected from the saphenous vein into heparinized tubes before treatment (time 0 min) and at 30 min, 6 h, 24 h and at intervals up to 135 days. Blood was withdrawn at similar time points from control mice. Another group of mice (n = 5 treatment; n = 5 control) was treated with 1500 mg kg−1 TOCP sc or the ethanol equivalent. Mice were sacrificed at 96 h post-dosing and cardially perfused to wash out blood before tissues were removed. Liver, spleen, fat (abdominal), skin, heart, muscle (quadriceps), testes, kidney, lung, small intestine, brain and plasma were collected from treated and control mice. Tissues were homogenized in 10 volumes of 50 mM potassium phosphate pH 7.4 containing 0.5% Tween-20. Homogenates were clarified by centrifugation. The supernatant was tested for acetylcholinesterase and butyrylcholinesterase activity.Functional observational battery
Mice were observed for the behavioral toxic signs described by McDaniel and Moser21 including posture, involuntary motor movements, tremors, seizures, convulsions, palpebral closure, reactivity to being handled, lacrimation, salivation, piloerection, gait, mobility, arousal, stereotypy, straub tail, vocalization, righting reflex, hyperactivity, and temperature.Temperature
Axial body temperature was measured with a digital thermometer, Thermalert model TH-5, attached to a surface Microprobe MT-D, type T thermocouple (Physitemp Instruments, Clifton, NJ). Temperature was recorded prior to challenge, at 5 min intervals for the first hour post-challenge, hourly through 8 h, and daily post dosing.Enzyme activity measurement
AChE activity in mouse plasma and selected tissues was assayed with 1 mM acetylthiocholine in 0.1 M potassium phosphate pH 7.0, 0.5 mM dithiobisnitrobenzoic acid, in the presence of 0.01 mM ethopropazine to inhibit BChE activity. BChE activity was assayed with 1 mM butyrylthiocholine in 0.1 M potassium phosphate pH 7.0, 0.5 mM dithiobisnitrobenzoic acid. Both assays were conducted at 25 °C. Three μl of plasma or 50 μl of clarified tissue homogenate supernatant were used in a 2 ml reaction volume in quartz cuvettes. The change in absorbance at 412 nm was recorded on a temperature-controlled spectrophotometer (Gilford Instrument Laboratories Inc., Oberlin, Ohio) interfaced to MacLab 200 (ADInstruments, Pty Ltd, Castle Hill, Australia) and a Macintosh computer. Activity was calculated in micromoles per minute from the extinction coefficient of 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 M−1 cm−1.22 A unit of AChE or BChE activity was defined as one micromole of substrate hydrolyzed per minute.
600 M−1 cm−1.22 A unit of AChE or BChE activity was defined as one micromole of substrate hydrolyzed per minute.
Nondenaturing gradient gel electrophoresis and staining for AChE activity
Gradient polyacrylamide gels (4–30%) were cast in a Hoefer SE600 gel apparatus (Hoefer, Holliston, MA) to make 15 × 11 cm gels, 0.75 mm thick. Plasma samples from control and treated mice were incubated with 0.1 mM iso-OMPA for 40 min to inhibit BChE. Following inhibition the plasma samples (7.5 μl per lane) were mixed with an equal volume of 50% glycerol, 0.1 M TrisCl pH 6.8, 0.1% bromophenol blue before loading on the gel. Mouse plasma that contained only BChE activity and no AChE activity was from AChE−/− mice.23 Mouse plasma that contained only AChE activity and no BChE activity was from BChE−/− mice.20 Electrophoresis was conducted at 250 V constant voltage for 20 h at 4 °C. Gels were soaked in 0.1 mM iso-OMPA in water for 30 min before they were stained for AChE activity by the method of Karnovsky and Roots.24 The staining solution contained 180 ml of 0.2 M maleic acid adjusted to pH 6 with 1 M NaOH, 30 ml of 0.03 M CuSO4, 30 ml of 5 mM potassium ferricyanide, 15 ml of 0.1 M sodium citrate, 30 ml water and 150 mg of acetylthiocholine iodide. The purpose of pretreating gels with iso-OMPA was to inhibit BChE. Since BChE hydrolyzes acetylthiocholine, reducing the intensity of BChE bands with iso-OMPA allowed better visualization of AChE bands. Gels were incubated overnight with gentle shaking. The reaction was stopped by washing the gels with water.Tissue sections
Tissues from mice treated with 1500 mg kg−1 TOCP for 4 days were prepared for sectioning (n = 5). Lungs were removed gently and inflated with Tissue-Tek O.C.T. through a tracheal cannula.25 The lung (inflated), heart, liver, brain, abdominal fat, quadriceps muscle, spleen, testes, kidney, and small intestine were embedded in O.C.T. compound on dry ice. The frozen tissues were sliced into 10 μm sections with a cryostat stick (Leica CM 1850) onto silanized slides. Apoptotic cells were identified by labeling the DNA breaks with fluorescent-tagged deoxyuridine triphosphate nucleotides in the TUNEL assay described below, and by staining for AChE activity.Double staining for AChE activity and TUNEL
The method for AChE activity staining is described by Karnovsky and Roots.24 All reagents were filtered through a 0.45 micron filter to remove particulates. Slides in an 8-slide glass staining jar were washed three times with 0.1 M maleic acid pH 6.0 and incubated in 1 mM iso-OMPA for 40 min to inhibit BChE. The iso-OMPA containing buffer was poured off and replaced with 75 ml of 0.12 M maleic acid pH 6 containing 5 mM sodium citrate, 3 mM copper sulfate, 0.5 mM potassium ferricyanide, and 2 mM acetylthiocholine. After 2 h incubation at room temperature, the slides were washed 3 times with 50 mM Tris–HCl pH 7.4. The reddish brown color of AChE positive areas was visualized with a Nikon Eclipse E800 microscope using a bright field source. The slides were counterstained for DNA breaks with fluorescent dUTP in a TUNEL assay. Images of cells stained for TUNEL were captured by a camera mounted on the Nikon Eclipse E800 microscope, using a fluorescent light source. The fluorescent light source consisted of an excitation band pass filter that provided 465–495 nm excitation light and an emission band pass filter that transmitted fluorescence at 515–555 nm.Terminal deoxynucleotidyl-transferase-mediated deoxyuridine triphosphate (dUTP) nick-end-labeling (TUNEL)
DNA fragments resulting from the apoptotic activation of intracellular endonucleases were detected in tissue sections by incorporation of fluorescein labeled dUTP using the In Situ Apoptosis Detection kit from Clontech, as described by the manufacturer. Briefly, sections were fixed in 4% paraformaldehyde for 30 min, washed twice with phosphate buffered saline, and permeabilized with permeabilization buffer for 5 min on ice. Sections were incubated with the TUNEL reaction mixture (consisting of Terminal deoxynucleotidyl-transferase enzyme and Labeling Safe Buffer) for three hours in a 37 °C humidified incubator. The labeling reaction was terminated by washing 3 times with phosphate buffered saline. Apoptotic cells were detected by fluorescence microscopy (Nikon, Eclipse E800) as described in the methods section on double staining for AChE activity and TUNEL. TUNEL positive cells were counted in 6 randomly selected fields at 100× magnification. Findings were compared between the study and control tissues.Statistical analysis
The results were expressed as mean values ± standard deviation. To evaluate statistical significance, a two-tailed t-test for dependent samples was used when the data were consistent with normality and wilcoxon signed rank test was used when the data were not normally distributed. The analysis used SPSS 16.0 software.Results
Minimal toxic signs in mice treated with tri-o-cresyl phosphate
Mice treated subcutaneously with a single dose of 1500 mg kg−1 TOCP had a decrease in body temperature of about 2 degrees at 6 h post dosing. Body temperature had returned to normal by 24 h post dosing. Body weight was unaffected by TOCP treatment. Mice euthanized on day 4 after treatment had enlarged spleens. However, mice euthanized on day 135 after treatment had normal size spleens.Plasma AChE and BChE activities after treatment with TOCP
Fig. 1 shows that plasma AChE activity was inhibited at 24 and 48 h post-dosing with 1500 mg kg−1 TOCP sc, but at no other times. The highest inhibition of plasma AChE activity occurred at 24 h post-dosing when AChE activity was inhibited 43% from 0.23 units per ml before treatment to 0.13 units per ml. AChE activity climbed back to normal over the next day. On day 3, AChE activity was 130% of normal. On day 4, AChE activity rose to 220% of the normal level. AChE activity remained elevated for 135 days, though the levels fluctuated from a high of about 220% to a low of about 130% of normal. The fluctuation in AChE activity may have been a result of the coprophagic behavior of mice, who eat their own feces and therefore potentially become re-exposed on a daily basis. The abnormally high plasma AChE activity had no toxic effects on the mice. Blood from control mice was withdrawn and tested for AChE and BChE activity at similar time points, to evaluate the possibility that the stress of the blood draw might affect activity. It was found that AChE and BChE activities in control mice were unaffected by repeated blood draws.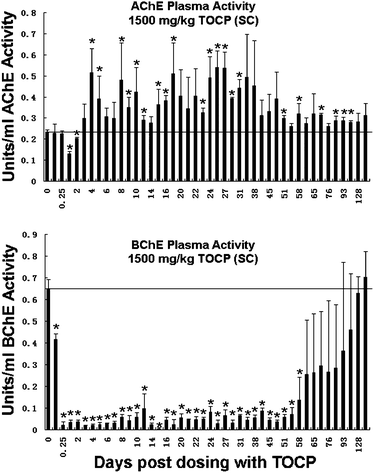 | ||
| Fig. 1 AChE and BChE activities in the plasma of mice (n = 4) treated with 1500 mg kg−1 TOCP. The x-axis shows days post dosing from 3 h to 135 days. The y-axis shows AChE activity (top panel) and BChE activity (lower panel) in units per ml. Error bars are ±standard deviation. The horizontal line represents the control activity before treatment. The symbol * indicates statistically significant differences in plasma AChE and BChE activities pre- and post-dosing for dependent samples (p < 0.05). ES1−/−BChE+/− mice. | ||
Fig. 1 shows that at 6 h (0.25 days) post-dosing plasma BChE activity was inhibited 97% from 0.65 units ml−1 before treatment to 0.02 units ml−1. Plasma BChE activity remained inhibited more than 90% until day 55. Thereafter, plasma BChE slowly increased. By day 128 BChE activity had returned to normal. BChE activity did not rise above baseline. These results confirm previous observations that inhibition of BChE activity is not associated with toxicity.
AChE activity stained polyacrylamide gel to visualize the size of AChE induced by TOCP
Plasma samples that were collected during the first ten days after dosing mice with 1500 mg kg−1 TOCP were analyzed on a non-denaturing gradient polyacrylamide gel stained for AChE activity (Fig. 2). Induced AChE activity migrated as a broad, fuzzy band in row 3 between tetrameric AChE in row 4 and dimeric AChE in row 2, suggesting that induced AChE is smaller than the 285 kDa AChE tetramer. Fuzzy bands of activity indicate heterogeneity in the structure of the AChE protein. The heterogeneity could come from partial proteolysis or from heterogeneity in the three asparagine-linked glycans of mouse AChE (Swiss Protein P21836; crystal structure PDB code 1MAA).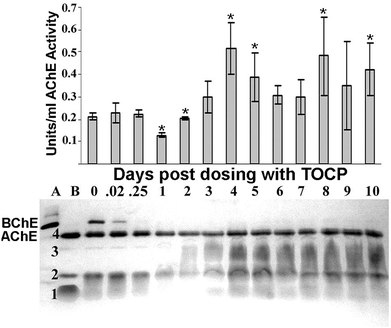 | ||
| Fig. 2 Visualization of AChE activity as a function of days post dosing with 1500 mg kg−1 TOCP. The top panel shows plasma AChE activity on days 0–10 post dosing. The bottom panel shows the molecular forms of AChE in the same plasma samples, as visualized on a non-denaturing gradient polyacrylamide gel stained for AChE activity. Plasma was loaded at 7.5 μl per lane. Lane (A) plasma from AChE knockout mouse (AChE−/−) demonstrating the location of a heavy BChE tetramer band. Lane (B) plasma from BChE knockout mouse (BChE−/−) demonstrating the location of the AChE bands. The band in row 4 is tetrameric AChE. The broad, fuzzy band in row 3 is an induced form of AChE. The band in row 2 could be dimeric AChE and that in row 1 could be monomeric AChE. The band for tetrameric BChE is present at time 0, but absent at 0.25 to 10 days post dosing, consistent with >90% inhibition of plasma BChE activity. Mouse genotype was ES1−/−BChE+/−. BChE activity is visible despite pretreatment of the gels with the BChE specific inhibitor iso-OMPA. This is consistent with a slow reversal of inhibition. The symbol * indicates statistically significant differences in plasma AChE and BChE activities pre- and post-dosing for dependent samples (p < 0.05). Error bars are ±standard deviation. | ||
Tissue AChE and BChE activities at 96 h after treatment with TOCP
Mice treated with 1500 mg kg−1 TOCP sc were sacrificed at 96 h, when plasma AChE achieved 220% of the normal activity. AChE and BChE activities were measured in liver, spleen, abdominal fat, skin, heart, quadriceps muscle, testis, kidney, lung, small intestine and brain tissues from control and TOCP treated mice (Fig. 3). AChE activity increased from 0.55 to 0.85 units g−1 in the spleens and from 0.14 to 0.22 units g−1 in the lungs of treated mice compared to control mice. No other tissue homogenate showed increased AChE activity compared to control. BChE activity was inhibited in liver, fat, muscle, testes, kidney, lung, and small intestine from TOCP treated mice when compared to control. For example, BChE activity in liver dropped from 1.6 to 0.8 units g−1 (50% inhibition). BChE activity in small intestine dropped from 1.8 to 0.6 units g−1 (67% inhibition).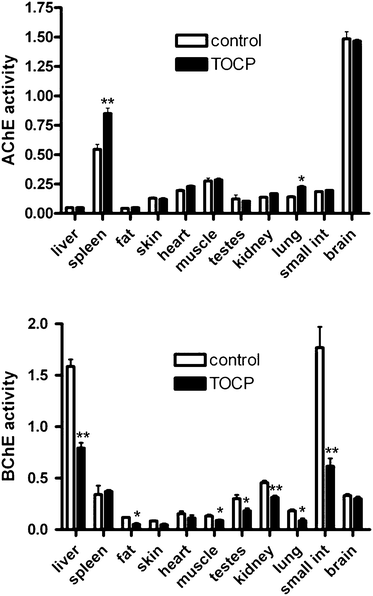 | ||
| Fig. 3 Tissue AChE and BChE activities in mice 96 h after treatment with 1500 mg kg−1 TOCP compared to control mice (n = 5 per group). AChE activity (top panel) and BChE activity (bottom panel) are expressed as micromoles per min per gram wet weight. Statistically significant differences between TOCP treated and control samples are indicated by * for p < 0.05, and by ** for p < 0.01. Mouse genotype was ES1−/−BChE+/−. Error bars are ±standard deviation. | ||
Apoptosis at 96 h after treatment with TOCP
Apoptosis was measured by counting TUNEL positive nuclei as an indicator of apoptotic activity in control and TOCP treated mouse tissue sections. Tissues examined for TUNEL positive cells included liver, spleen, fat, heart, muscle, testes, kidney, lung, small intestine, and brain. An increase in the number of apoptotic cells was seen in lung sections from TOCP treated mice compared to control mice (Fig. 4). There was no significant difference in the number of apoptotic cells in spleen and other tissues.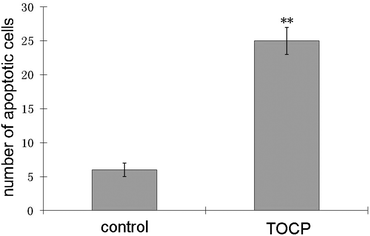 | ||
| Fig. 4 Quantification of TUNEL-positive cells in lung sections from mice treated with a single dose of 1500 mg kg−1 TOCP sc compared to control mice. Lungs were obtained on day 4 post-dosing. TUNEL-positive cells were defined by counting the number of green fluorescent nuclei in 6 randomly chosen fields. Statistically significant differences are indicated by ** for p < 0.01. Error bars are ±standard deviation. | ||
AChE activity associated with apoptosis
It was of interest to determine whether apoptotic cells had high levels of AChE activity. For this purpose, tissue sections were stained for AChE activity followed by TUNEL staining of the same sections to identify genomic DNA fragmentation. As shown in Fig. 5, cells that stained heavily for AChE activity were also brightly fluorescent in the TUNEL assay. All TUNEL-positive cells were positive for AChE activity, but only 50% of areas that stained intensely for AChE activity were TUNEL-positive. It is apparent that apoptotic cells are associated with high levels of AChE activity. That is, toxicants that induce apoptosis in the organs of a living mouse simultaneously induce excess AChE activity in apoptotic cells. Organophosphorus agents inhibit AChE immediately after exposure, but with time an excess of AChE activity appears in the plasma. The excess plasma AChE may be released from cells undergoing apoptosis.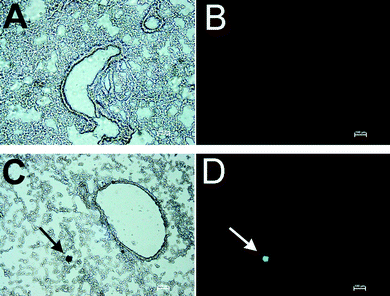 | ||
| Fig. 5 Double staining for AChE activity and TUNEL assay. Mice treated with 1500 mg kg−1 TOCP and control mice were sacrificed at 4 days post dosing. A: Section of inflated lung from control mouse stained for AChE activity; dark brown areas are positive for AChE activity. B: The section in A counterstained for fragmentation of genomic DNA with the TUNEL assay shows no fluorescent cells. C: Section of inflated lung from a TOCP treated mouse stained for AChE activity; the arrow points to a cell that stains heavily for AChE activity. D: The section in C counterstained for fragmentation of genomic DNA with the TUNEL assay; the arrow points to the green fluorescent cell that is positive for genomic DNA fragmentation. Scale bar, 100 μm. | ||
Discussion
Long lasting effect of a single dose of TOCP
Mice treated with a single high dose (1500 mg kg−1) of TOCP had elevated plasma AChE activity and reduced plasma BChE activity for 2 to 4 months. The long duration of action suggests TOCP was stored in the body and slowly released to the liver where cytochrome P450 enzymes metabolized it into the toxic cresyl saligenin phosphate.14 See Fig. 6 for structures of TOCP and cresyl saligenin phosphate. TOCP is highly lipophilic, so that long term storage in fat depots would be expected. This is consistent with the slow release hypothesis for the long lasting effect from a single dose of TOCP. The toxic metabolite inhibited BChE more extensively than AChE, consistent with the finding that cresyl saligenin phosphate is 100-fold more reactive with human BChE than with human AChE.16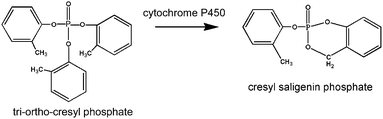 | ||
| Fig. 6 Structures of TOCP and cresyl saligenin phosphate. | ||
Mice treated with 30 mg kg−1 TOCP sc showed a similar pattern of plasma AChE activity induction, which started on day 4 after treatment.2 However the 30 mg kg−1 dose did not inhibit plasma AChE activity. BChE activity was significantly reduced by 6 h post dosing with a return to baseline levels by 21 days post dosing.
Effect of mouse genotype on induction of abnormally high levels of AChE
The present work used ES1−/−BChE+/− mice that were completely deficient in plasma carboxylesterase and partially deficient in BChE. Carboxylesterase and butyrylcholinesterase deficient mice were used as models for humans because humans have no plasma carboxylesterase26 and 1 out of 4 humans has a BChE deficient genotype. In a previous report we used wild-type mice.2 Both genotypes responded to OP treatment with an initial phase in which AChE activity was inhibited followed by induction of plasma AChE activity to levels above baseline. This suggests that plasma carboxylesterase and butyrylcholinesterase have no role in induction of excess AChE activity.Excess AChE in plasma is a biomarker of toxicity
Mice treated with TOCP had minimal behavioral signs of toxicity, yet they had abnormally high plasma AChE activity lasting months. We demonstrated that the TOCP treated mice had more apoptotic cells in lung tissue than were found in untreated control mice. We also demonstrated that apoptotic cells were associated with high levels of AChE activity. It follows that the increase in AChE activity observed in plasma comes from the apoptotic cells. We conclude that excess plasma AChE activity is associated with apoptotic cells and is an indicator of exposure to toxicants that induce apoptosis.Mice and other rodents have significant amounts of AChE in plasma, but humans have almost none. In healthy humans the concentration of AChE in plasma is about 8 ng ml−1, a value 400-fold less than that of butyrylcholinesterase.27 Alzheimer disease patients have 20% higher AChE activity in plasma compared to age and gender-matched controls, but no change in BChE levels.28 The increased AChE in plasma could be the consequence of apoptotic cells releasing AChE into the circulation.
Tissue source of excess AChE in plasma
The lungs of TOCP treated mice had 1.5-fold higher AChE activity and 4-fold more apoptotic cells than the lungs of control mice. Studies with radiolabeled TOCP have shown that high levels of TOCP or its metabolites accumulate in the lungs of mice and rats.29,30 It was suggested that covalent binding of TOCP or its reactive metabolites to alveolar macromolecules may explain the high concentration of radiolabel in mouse lungs.30 Covalent binding of toxicant to macromolecules could damage the lung, thus explaining the excess number of apoptotic cells that we observed in the lung. Apoptotic cells in the lung could be a source of excess AChE in the plasma of TOCP treated mice.Role of AChE in apoptosis
It has been proposed that the role of AChE in apoptosis is to facilitate the interaction of proteins that constitute the apoptosome31 where AChE serves an adhesive function. Recruitment of AChE to the apoptosome completes the mutiprotein complex. Subsequently procaspases are activated to functional proteases, thus initiating programmed cell death. Inhibition of AChE activity with physostigmine or BW284c51 did not prevent apoptosis in Jurkat and HL-60 cells,32 suggesting that the adhesive property of the AChE protein rather than AChE catalytic activity is important for apoptosis.Conclusions
Exposure of mice to TOCP results in an increase in apoptotic activity. Cells staining for apoptotic activity also stain for increased AChE activity. An overall increase in plasma AChE activity correlates with the increased apoptotic activity in TOCP treated mice. These observations suggest that increased AChE activity is a biomarker for apoptotic activity.Conflicts of interest statement
The authors declare no conflict of interest.Funding
WJ was supported by a fellowship from the China Scholarship Council and a Program of Excellence Graduate Assistantship from the University of Nebraska Medical Center.Abbreviations
| AChE | acetylcholinesterase |
| BChE | butyrylcholinesterase |
| iso-OMPA | tetraisopropyl pyrophosphoramide |
| OP | organophosphorus agent |
| TOCP | tri-o-cresyl phosphate |
| TUNEL | terminal deoxyribonucleotidyl transferase mediated nick end labeling |
Notes and references
- L. W. Harris, H. I. Yamamura and J. H. Fleisher, De novo synthesis of acetylcholinesterase in guinea pig retina after inhibition by pinacolyl methylphosphonofluoridate, Biochem. Pharmacol., 1971, 20, 2927–2930 CrossRef CAS.
- E. G. Duysen and O. Lockridge, Induction of plasma acetylcholinesterase activity in mice challenged with organophosphorus poisons, Toxicol. Appl. Pharmacol., 2011, 255, 214–220 CrossRef CAS.
- S. Chiappa, S. Padilla, C. Koenigsberger, V. Moser and S. Brimijoin, Slow accumulation of acetylcholinesterase in rat brain during enzyme inhibition by repeated dosing with chlorpyrifos, Biochem. Pharmacol., 1995, 49, 955–963 CrossRef CAS.
- R. Trevisan, M. Uliano-Silva, P. Pandolfo, J. L. Franco, P. S. Brocardo, A. R. Santos, M. Farina, A. L. Rodrigues, R. N. Takahashi and A. L. Dafre, Antioxidant and acetylcholinesterase response to repeated malathion exposure in rat cerebral cortex and hippocampus, Basic Clin. Pharmacol. Toxicol., 2008, 102, 365–369 CrossRef CAS.
- I. Bansal, C. K. Waghmare, T. Anand, A. K. Gupta and B. K. Bhattacharya, Differential mRNA expression of acetylcholinesterase in the central nervous system of rats with acute and chronic exposure of sarin & physostigmine, J. Appl. Toxicol., 2009, 29, 386–394 CrossRef CAS.
- K. Carlson, B. S. Jortner and M. Ehrich, Organophosphorus compound-induced apoptosis in SH-SY5Y human neuroblastoma cells, Toxicol. Appl. Pharmacol., 2000, 168, 102–113 CrossRef CAS.
- A. M. Saleh, C. Vijayasarathy, L. Masoud, L. Kumar, A. Shahin and A. Kambal, Paraoxon induces apoptosis in EL4 cells via activation of mitochondrial pathways, Toxicol. Appl. Pharmacol., 2003, 190, 47–57 CrossRef CAS.
- A. Caughlan, K. Newhouse, U. Namgung and Z. Xia, Chlorpyrifos induces apoptosis in rat cortical neurons that is regulated by a balance between p38 and ERK/JNK MAP kinases, Toxicol. Sci., 2004, 78, 125–134 CrossRef CAS.
- Q. Li, M. Kobayashi and T. Kawada, Chlorpyrifos induces apoptosis in human T cells, Toxicology, 2009, 255, 53–57 CrossRef CAS.
- M. G. Aluigi, C. Guida and C. Falugi, Apoptosis as a specific biomarker of diazinon toxicity in NTera2-D1 cells, Chem.-Biol. Interact., 2010, 187, 299–303 CrossRef CAS.
- M. P. Kashyap, A. K. Singh, V. Kumar, V. K. Tripathi, R. K. Srivastava, M. Agrawal, V. K. Khanna, S. Yadav, S. K. Jain and A. B. Pant, Monocrotophos induced apoptosis in PC12 cells: role of xenobiotic metabolizing cytochrome P450s, PLoS One, 2011, 6, e17757 CAS.
- X. J. Zhang, L. Yang, Q. Zhao, J. P. Caen, H. Y. He, Q. H. Jin, L. H. Guo, M. Alemany, L. Y. Zhang and Y. F. Shi, Induction of acetylcholinesterase expression during apoptosis in various cell types, Cell Death Differ., 2002, 9, 790–800 CrossRef CAS.
- C. Winder and J. C. Balouet, The toxicity of commercial jet oils, Environ. Res., 2002, 89, 146–164 CrossRef CAS.
- M. Eto, J. E. Casida and T. Eto, Hydroxylation and cyclization reactions involved in the metabolism of tri-o-cresyl phosphate, Biochem. Pharmacol., 1962, 11, 337–352 CrossRef CAS.
- P. Glynn, A mechanism for organophosphate-induced delayed neuropathy, Toxicol. Lett., 2006, 162, 94–97 CrossRef CAS.
- E. Carletti, L. M. Schopfer, J. P. Colletier, M. T. Froment, F. Nachon, M. Weik, O. Lockridge and P. Masson, Reaction of Cresyl Saligenin phosphate, the organophosphorus agent implicated in aerotoxic syndrome, with human cholinesterases: mechanistic studies employing kinetics, mass spectrometry, and x-ray structure analysis, Chem. Res. Toxicol., 2011, 24, 797–808 CrossRef CAS.
- M. Liyasova, B. Li, L. M. Schopfer, F. Nachon, P. Masson, C. E. Furlong and O. Lockridge, Exposure to tri-o-cresyl phosphate detected in jet airplane passengers, Toxicol. Appl. Pharmacol., 2011, 256, 337–347 CrossRef CAS.
- M. B. Abou-Donia and D. M. Lapadula, Mechanisms of organophosphorus ester-induced delayed neurotoxicity: type I and type II, Annu. Rev. Pharmacol., 1990, 30, 405–440 CrossRef CAS.
- E. G. Duysen, F. Koentgen, G. R. Williams, C. M. Timperley, L. M. Schopfer, D. M. Cerasoli and O. Lockridge, Production of ES1 plasma carboxylesterase knockout mice for toxicity studies, Chem. Res. Toxicol., 2011, 24, 1891–1898 CrossRef CAS.
- B. Li, E. G. Duysen, M. Carlson and O. Lockridge, The butyrylcholinesterase knockout mouse as a model for human butyrylcholinesterase deficiency, J. Pharmacol. Exp. Ther., 2008, 324, 1146–1154 CrossRef CAS.
- K. L. McDaniel and V. C. Moser, Utility of a neurobehavioral screening battery for differentiating the effects of two pyrethroids, permethrin and cypermethrin, Neurotoxicol. Teratol., 1993, 15, 71–83 CrossRef CAS.
- G. L. Ellman, K. D. Courtney, V. Andres, Jr. and R. M. Featherstone, A new and rapid colorimetric determination of acetylcholinesterase activity, Biochem. Pharmacol., 1961, 7, 88–95 CrossRef CAS.
- W. Xie, J. A. Stribley, A. Chatonnet, P. J. Wilder, A. Rizzino, R. D. McComb, P. Taylor, S. H. Hinrichs and O. Lockridge, Postnatal developmental delay and supersensitivity to organophosphate in gene-targeted mice lacking acetylcholinesterase, J. Pharmacol. Exp. Ther., 2000, 293, 896–902 CAS.
- M. J. Karnovsky and L. Roots, A “direct-coloring” thiocholine method for cholinesterases, J. Histochem. Cytochem., 1964, 12, 219–221 CrossRef CAS.
- J. K. Myung, G. Choe, D. H. Chung, J. W. Seo, S. Jheon, C. T. Lee and J. H. Chung, A simple inflation method for frozen section diagnosis of minute precancerous lesions of the lung, Lung Cancer, 2008, 59, 198–202 CrossRef.
- B. Li, M. Sedlacek, I. Manoharan, R. Boopathy, E. G. Duysen, P. Masson and O. Lockridge, Butyrylcholinesterase, paraoxonase, and albumin esterase, but not carboxylesterase, are present in human plasma, Biochem. Pharmacol., 2005, 70, 1673–1684 CrossRef CAS.
- S. Brimijoin and P. Hammond, Butyrylcholinesterase in human brain and acetylcholinesterase in human plasma: trace enzymes measured by two-site immunoassay, J. Neurochem., 1988, 51, 1227–1231 CrossRef CAS.
- M. S. Garcia-Ayllon, I. Riba-Llena, C. Serra-Basante, J. Alom, R. Boopathy and J. Saez-Valero, Altered levels of acetylcholinesterase in Alzheimer plasma, PLoS One, 2010, 5, e8701 Search PubMed.
- M. B. Abou-Donia, A. A. Nomeir, J. H. Bower and H. A. Makkawy, Absorption, distribution, excretion and metabolism of a single oral dose of [14C]tri-o-cresyl phosphate (TOCP) in the male rat, Toxicology, 1990, 65, 61–74 CrossRef CAS.
- A. E. Ahmed, S. Jacob, S. Soliman, N. Ahmed, K. Osman, J. P. Loh and N. Romero, Whole-body autoradiographic disposition, elimination and placental transport of [14C]tri-o-cresyl phosphate in mice, J. Appl. Toxicol., 1993, 13, 259–267 CrossRef CAS.
- S. E. Park, N. D. Kim and Y. H. Yoo, Acetylcholinesterase plays a pivotal role in apoptosome formation, Cancer Res., 2004, 64, 2652–2655 CrossRef CAS.
- X. Huang, B. Lee, G. Johnson, J. Naleway, A. Guzikowski, W. Dai and Z. Darzynkiewicz, Novel assay utilizing fluorochrome-tagged physostigmine (Ph-F) to in situ detect active acetylcholinesterase (AChE) induced during apoptosis, Cell Cycle, 2005, 4, 140–147 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |