In vitro and in vivo studies into the biological activities of 1,10-phenanthroline, 1,10-phenanthroline-5,6-dione and its copper(II) and silver(I) complexes
Malachy
McCann
*a,
André L. S.
Santos
b,
Bianca A.
da Silva
b,
Maria Teresa V.
Romanos
c,
Alexandre S.
Pyrrho
d,
Michael
Devereux
e,
Kevin
Kavanagh
f,
Iduna
Fichtner
g and
Andrew
Kellett
h
aChemistry Department, National University of Ireland, Maynooth, Kildare, Ireland. E-mail: malachy.mccann@nuim.ie; Tel: +353 (0)1 7083767
bDepartamento de Microbiologia Geral, Instituto de Microbiologia Prof. Paulo de Góes (IMPPG), Universidade Federal do Rio de Janeiro (UFRJ), Brazil
cDepartamento de Virologia, IMPPG, UFRJ, Brazil
dDepartamento de Análises Clínicas e Toxicológicas, Faculdade de Farmácia, UFRJ, Brazil
eThe Inorganic Pharmaceutical and Biomimetic Research Centre, Focas Research Institute, Dublin Institute of Technology, Camden Row, Dublin 8, Ireland
fMedical Mycology Unit, NICB, Department of Biology, National University of Ireland, Maynooth, Co. Kildare, Ireland
gMax Delbrück Center for Molecular Medicine, Experimental Pharmacology, Robert-Rössle-Str. 10, 13125 Berlin, Germany
hSchool of Chemical Sciences and National Institute of Cellular Biotechnology, Dublin City University, Glasnevin, Dublin 9, Ireland
First published on 16th May 2012
Abstract
1,10-Phenanthroline (phen, 5), 1,10-phenanthroline-5,6-dione (phendione, 6), [Cu(phendione)3](ClO4)2·4H2O (12) and [Ag(phendione)2]ClO4 (13) are highly active, in vitro, against a range of normal and cancerous mammalian cells, fungal and insect cell lines, with the metal complexes offering a clear enhancement in activity. Cytoselectivity was not observed between the tumorigenic and non-tumorigenic mammalian lines. In in vivo tests, using Galleria mellonella and Swiss mice, all four compounds were well tolerated in comparison to the clinical agent, cisplatin. In addition, blood samples taken from the Swiss mice showed that the levels of the hepatic enzymes, aspartate aminotransferase (AST) and alanine aminotransferase (ALT), remained unaffected. Immunocompromised nude mice showed a much lower tolerance to 13 and, subsequently, when these mice were implanted with Hep-G2 (hepatic) and HCT-8 (colon) human-derived tumors, there was no influence on tumor growth.
Introduction
Metal complexes of nitrogen-substituted, phenanthrene-based ligands have shown significant potential as broad-spectrum agents capable of eliciting cytotoxicity toward diseases and infections manifested by cancer,1–8 viruses,9–11 bacteria6,12–15 and fungi.16–18 The planar, aromatic phenanthrene molecule (1) (Fig. 1) is widely found in nature and forms the backbone of many natural and semi-synthetic opiates, including morphine, codeine and naloxone. A selection of heteroaromatic derivatives of phenanthrene (2–7 and 9–11) is shown in Fig. 1, and although the quinone derivatised compounds (4, 6 and 8) experience loss of aromaticity at the central arene ring, they do retain their planar structure. The N-substituted organic molecule, 9-phenanthridine (2), has been compared structurally to the well-known polycyclic DNA intercalator, acridine (8),19–21 and studies of the nucleotide binding capability of cationic phenanthridinium derivatives have already been reported.22 | ||
| Fig. 1 Structures of selected compounds containing the phenanthrene-type backbone. | ||
Early studies on the antibacterial properties of the metal-free compounds, 1–7, revealed that inclusion of N-atoms in the phenanthrene ring generally increased antimicrobial activity.15 Interestingly, the quinone (dione) derivatives, 4, 6 and 7, were found to increase antimicrobial efficacy further and were highly effective against the more resilient Gram-negative bacteria. In that study, of all compounds tested (1–7), 1,10-phenanthroline-5,6-quinone (phendione) (6) was found to be the most effective. Indeed, when metal-free N,N′-chelating bases are found to be bioactive, it is assumed that the sequestering of trace metals is involved and that the resulting metal complexes are the actual active species.13 Thus, one possible explanation for the enhanced activity exhibited by 4 and 7 could relate to their ability to form metal complexes via cellular O,O′ (4) or N,O (7) metal chelation. The relationship between biological efficacy and N,N′-chelation within this class of molecule was further strengthened through our previous observations on the control of Candida albicans fungal isolates by the metal-free compounds, 3, 5 and 9.16 At a concentration of 20 μg mL−1, 1,10-phenanthroline (phen) (5) showed potent antifungal bioactivity (0% cell growth during 24 h) while the non-chelating isomers, 3 and 9, were largely inactive (93% and 99% cell growth, respectively). In the same study, Ag(I), Cu(II) and Mn(II) ternary complexes, comprising 5 and malonate ligands, were also found to be highly toxic toward C. albicans. The success of metal-free phen along with these Ag(I), Cu(II) and Mn(II) ternary complexes is attributed to their ability to induce extensive changes to the internal structure of yeast cells, which included, retraction of the cytoplasm, nuclear fragmentation and disruption of mitochondrial function leading to apoptosis.17,24 Phen (5) is also fungicidal against the multi-resistant filamentous fungus belonging to the Pseudallescheria boydii/Scedosporium apiospermum complex, with the antifungal activity being directly dependent on both cell density and phen concentration.25
The application of metal complexes of phen (5) and its substituted quinone and quinoxaline derivatives (6, 10 and 11) as novel antitumor agents has stimulated intensive interest, particularly within the past two decades.1,8,26–35 The application of such agents for the control of cancer, however, must be considered in conjunction with their ability to perturb the natural intestinal microbial flora that is necessary for, among other things, resistance to invasive pathogenic bacteria. Recently, Pt(II) complexes of general formula [PtLCl2] (L = 6 or 10) were reported as broad-spectrum antitumor agents which, interestingly, exhibited negligible antibacterial activity (Bacillus subtilis, Escherichia coli; MIC > 200 μg mL−1).6 However, it must be highlighted that cisplatin alone exhibits moderate antibacterial activity and this has been shown to potentiate the virulence of Candida cells and enhance the risk of systemic candidiasis, which can ultimately lead to fatalities amongst cancer patients.36 Thus, while antitumor complexes exhibiting strong cytotoxicity toward a range of pathogens may be viewed as less obvious cancer drug candidates, it may be worth considering the ability of these compounds to control such virulent pathogens in conjunction with their propensity to treat cancer, particularly where immunosuppressive complications have arisen.
Metal complexes containing phen-type ligands represent a class of compound that are entirely different to the current Pt(II) clinical anticancer drugs for a number of reasons. (i) The planar, heteroaromatic phen ligand (5) and particularly its quinoxaline derivatives, 10 and 11, facilitate DNA binding via three distinctive modes: (a) hydrophobic interactions in the minor groove, (b) partial intercalation of the phen ligand into the helix in the major groove and (c) π-stacking by metallo-intercalators (e.g. phenazine) between base-paired regions.26,27 (ii) It has been demonstrated that metal coordination complexes comprising phen-type ligands enhance the in vitro activity of the oncogene, p53, a vital tumor-suppressor gene which functions by inducing apoptosis and preventing gene amplification and which is found mutated in many forms of human cancer.37 Additionally, owing to the low lying π* N aromatic orbitals present on phen (or derivatives thereof), bis– and tris–phen coordination complexes are characterised spectroscopically by intense metal-to-ligand charge-transfer transitions (MLCT),38 consequently leading to their potential application as photochemical redox or photodynamic therapeutic (PDT) agents.39–43
In our studies to date, we have prepared and screened a wide spectrum of metal-based agents for their antimicrobial and anticancer activities.16,17,23,24,32,44–48 In the majority of cases, it was observed that Cu2+ and Ag+ complexes containing phendione (6) ligands exhibit excellent biological activity and represent a class of DNA-targeting compounds capable of inhibiting nucleotide synthesis.5,16 As a logical progression of these studies, and given that very few reports exist in the literature on the in vivo potential of metal-phen adducts, the present paper details both the in vitro and in vivo chemotherapeutic potential, drug toxicity profiles and mechanistic aspects of phen (5), phendione (6) and Cu2+ and Ag+ complexes of 6, namely [Cu(phendione)3](ClO4)2·4H2O (12)23 and [Ag(phendione)2]ClO4 (13)23 (Fig. 2). Specifically, we report the in vitro cytotoxicity against mammalian tumor and non-tumor lines, macrophages, insect and fungal cell lines, the in vivo tolerance of larvae of the insect Galleria mellonella, the in vivo tolerance of 13 within Swiss and nude mice, and nude mouse xenograft studies of 13 against the human-derived, implanted tumors HCT-8 (colon) and Hep-G2 (kidney).
2 (12)23,49 and [Ag(phendione)2]ClO4 (13).23](/image/article/2012/TX/c2tx00010e/c2tx00010e-f2.gif)
Experimental
Synthesis
Chemicals were purchased from Sigma-Aldrich Ireland and used as received. 1,10-Phenanthroline-5,6-quinone (phendione (6)),50 [Cu(phendione)3](ClO4)2·4H2O and [Ag(phendione)2]ClO4 were prepared in accordance with the literature methods.23G. Mellonella toxicity
G. mellonella larvae in the 6th developmental stage were used to determine the in vivo cytotoxicity of 5, 6, 12, 13 and cisplatin. Thirty healthy larvae between 0.200–0.400 g in weight and with no cuticle discolouration were used for each experiment. Fresh solutions of the test compounds were prepared immediately prior to testing under sterile conditions. Each compound (0.05 g) was dissolved in DMSO (1 cm3) and added to sterile water (9 cm3) to give a stock solution of concentration 5000 mg cm−3. Each compound was tested across the concentration range 5000–200 μg cm−3. Test solutions (20 μL) were administered to the larvae by injection directly into the haemocoel through the last pro-leg. The base of the pro-leg can be opened by applying gentle pressure to the sides of the larvae and this aperture will re-seal after removal of the syringe without leaving a scar. Larvae were placed in sterile Petri dishes and incubated at 30 °C for 72 h. The survival of the larvae was monitored every 24 h. Death was assessed by the lack of movement in response to stimulus together with discolouration of the cuticle. Three controls were employed in all assays. The first consisted of untouched larvae maintained at the same temperature as the test larvae. The second was larvae with the pro-leg pierced with an inoculation needle but no solution injected. The third control was larvae that were inoculated with 20 μL of sterile distilled water.Antimicrobial assessment
Pseudallescheria boydii (RKI07_0416) belongs to the clade 4 of the Pseudallescheria boydii/Scedosporium apiospermum complex as previously proposed by Gilgado and co-workers.51 The fungus was grown on Sabouraud-dextrose agar (containing 2% glucose, 1% peptone, 0.5% yeast extract and 2% agar) plates at 25 °C for 7 days. Conidia were harvested and washed with sterile 10 mM phosphate-buffered saline (PBS, pH 7.2). The cells were separated by gauze filtration, then collected by centrifugation and washed three times in PBS.25 Cell density was estimated by counting the conidia in a Neubauer chamber.Cytotoxicity assays
In order to determine the cytotoxicity of the phen (5) and its derivatives, different concentrations of each compound (ranging from 10 to 0.0001 μg mL−1) were placed in contact with the cell cultures and incubated at 37 °C in a 5% CO2 atmosphere for 24 h, except for insect cell line in which the incubation was at 27 °C. After that, cellular viability was evaluated by the neutral red dye-uptake method.52 Briefly, cells were incubated in the presence of 0.01% neutral red solution for 3 h at 37 °C in a 5% CO2 atmosphere. The medium was then removed and the cells were fixed with 4% formalin in PBS (pH 7.2). The dye incorporated by the viable cells was eluted using a mixture of methanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acetic acid:water (50:1:49), and the dye uptake was determined by measuring the optical density (OD) of the eluate at 490 nm in an automatic spectrophotometer (ELx800TM-Bio-TeK Instruments, Inc.). The 50% cytotoxic concentration (CC50) was defined as the compound concentration which caused a 50% reduction in the number of viable cells.
:acetic acid:water (50:1:49), and the dye uptake was determined by measuring the optical density (OD) of the eluate at 490 nm in an automatic spectrophotometer (ELx800TM-Bio-TeK Instruments, Inc.). The 50% cytotoxic concentration (CC50) was defined as the compound concentration which caused a 50% reduction in the number of viable cells.
Acute toxicity testing in Swiss mice
All animal experiments were performed according to the Brazilian Animal Protection Laws and with approval from the local responsible authorities at the Instituto de Microbiologia Paulo de Góes (IMPG), Universidade Federal do Rio de Janeiro. The acute toxicity tests were performed with albino Swiss mice (females) weighing 20 ± 2 g, which were obtained from Institute of Microbiology Prof. Paulo de Góes-UFRJ facilities. For each inhibitor concentration tested five mice were used, having been applied volumes of 0.1 ml containing compounds in concentrations ranging from 15 to 450 mg kg−1 administered by intraperitoneal route. The control group received no injection and one group of animals received injections containing DMSO 10% (concentration of DMSO used to dilute the drugs). After the injections, the animals were observed during the first 5 h and thereafter every 6 to 12 h until a total of 168 h (7 days).Chronic toxicity testing in Swiss mice
Chronic toxicity tests were performed using an inhibitor concentration equivalent to 45 mg kg−1. The intraperitoneal injections were conducted daily for 5 consecutive days. All compounds were diluted in DMSO (10%) and the volume injected was 0.1 ml. A control was carried out with a mice group that did not receive an injection and another group that were injected for 5 consecutive days only with DMSO 10%. After receiving all of the injections the animals were monitored for 7 days and blood samples were collected and processed at the clinical laboratory of the Faculty of Pharmacy, UFRJ. Sera sample were used to determine the levels of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) enzymes, which are used as markers of liver integrity (Labtest, Lagoa Santa, MG, Brazil).In vivo xenograft experiments using nude mice
All animal experiments were performed according to the German Animal Protection Law and with approval from the local responsible authorities. Crl : NMRI.nu/nu mice were purchased from Charles River (Sulzfeld, Germany) and maintained under standardized (22 ± 2 °C, 50 ± 10% relative humidity, 12 h light–dark-rhythm) and pathogen-free conditions. For the approximate tolerability test, tumor-free male mice were administered once i.p. with complex 13. Behavioural anomalies, body weight, body weight change (BWC, in relation to the first measurement in %) and mortality were registered. In the therapeutic experiments, the mice received, at day zero, 107 Hep-G2 (human hepatocellular carcinoma or HCT-8 (human colon carcinoma) cells subcutaneously into the left flank. When tumors were palpable (day 22 for Hep G2, day 8 for HCT-8) the mice were randomized to the corresponding treatment groups. Treatment was performed in a q4dx3 schedule i.p. Tumor sizes were measured twice per week with a caliper and tumor volumes were calculated according to length × width2/0.5. Treated to control (T/C) values of mean tumor volumes were calculated at each measurement day and the optimum (lowest) value was recorded. Additionally, body weight was determined twice per week as an estimation of tolerability. Blood samples from 5 mice per group were taken from the retroorbital bulbus 4 days after initiation of treatment and blood parameters (WBC white blood cells, throm. thrombocytes) were determined with a Coulter counter. The experiments were finished for ethical reasons at day 45 or 22, respectively.Results and discussion
In almost every case, metal-free phendione (6) was considerably more cytotoxic than metal-free phen (5) (Table 1). When viewed in terms of micromolar concentrations, a further notable enhancement in activity was observed on progressing from 6 to the Cu2+ complex, 12. Furthermore, with the exception of C. albicans, the Ag+ complex, 13, was less potent than 12. For most of the cell lines, 13 was more active than metal-free 6. The general order of activity for the four test compounds is as follows: 12 > 13 > 6 > 5. The test compounds showed higher activities toward the human derived tumor and normal cell lines and fungal cell lines when compared to murine (except S49 and S180), simian and insect-derived cell lines. From the IC50 values shown in Table 1 it is evident that peritoneal mouse macrophages (MΦ) and the insect cell line from Aedes albopictus (C6/36) (both >10(55)) are, in general, more tolerant to all of the test compounds.| Cell lineages | Cytotoxicity IC50 μg mL−1 (μM) | |||
|---|---|---|---|---|
| Tested compounds | ||||
| 5 | 6 | 12 | 13 | |
| a IC50 μg mL−1 (μM) values for all cells except C. albicans where the MIC100 μg mL−1 (μM) values are quoted.23 Cell lineages: Vero (African green monkey kidney cell), MA-104 (kidney embryonic cell of Rhesus monkey), LLC-MK2 (kidney cell of Rhesus monkey), CHO (Chinese hamster ovary cells), S49 (mouse lymphoma cells), S180 (mouse cancer cells), RAW 264.7 (murine macrophages), MΦ (peritoneal mouse macrophages), HEp-2 (human larynx carcinoma cells), A549 (human Caucasian lung carcinoma), DLKP cells (human lung carcinoma cell line), A-498 (human renal carcinoma cell line), HK-2 (human renal normal cell line), Hep-G2 (human liver carcinoma cell line), Chang (human liver normal cell line), MRC-5 (human lung fibroblast), C6/36 (insect cell line from Aedes albopictus). b n.t. = not tested. | ||||
| Vero | >10 (55) | 6.6 (31.4) | 6.5 (6.7) | >10 (15.9) |
| MA-104 | >10 (55) | 3.9 (18.6) | 4.4 (4.6) | 7.3 (11.6) |
| LLC-MK2 | >10 (55) | 7.4 (35.2)) | 6.8 (7.0) | 9.1 (14.5) |
| CHO | >10 (55) | 7.9 (37.6) | 9.2 (9.5) | >10 (15.9) |
| S497 | n.t. | 0.0118 (0.056) | n.t. | n.t. |
| S1807 | n.t. | 0.0088 (0.042) | n.t. | n.t. |
| RAW | >10 (55) | 5.6 (26.6) | 6.1 (6.3) | >10 (15.9) |
| MΦ | >10 (55) | >10 (48) | >10 (10) | >10 (15.9) |
| MRC-5 | >10 (55) | 7.6 (36.2) | 7.6 (7.9) | 7.4 (11.8) |
| HEp-2 | >10 (55) | 8.8 (41.9) | >10 (10) | >10 (15.9) |
| A549 | >10 (55) | 8.5 (40.4) | 8.2 (8.5) | >10 (15.9) |
| DLKP23 | 0.35 (1.9) | 0.008 (0.04) | n.t. | 0.025 (0.04) |
| A49824 | 1.05 (5.8) | 0.88 (4.2) | 0.85 (0.88) | 0.88 (1.4) |
| HK-224 | 0.94 (5.2) | 0.15 (0.7) | 0.48 (0.5) | 0.50 (0.8) |
| Hep-G224 | 0.74 (4.1) | 0.29 (1.4) | 0.75 (0.78) | 0.54 (0.86) |
| Chang24 | 1.32 (7.3) | 0.08 (0.4) | 0.19 (0.2) | 0.19 (0.3) |
| C6/36 | >10 (55) | >10 (48) | >10 (10) | >10 (15.9) |
| P. boydii conidia | 0.473 (2.62) | 0.032 (0.15) | 0.096 (0.1) | 0.116 (0.18) |
| C. albicans yeasts23 | 2.5 (13.9) | 0.6 (2.9) | 1.3 (1.3) | 0.3 (0.48) |
The increasing prevalence of fungal infections, especially hospital-acquired infections and infections in immunocompromised patients, has highlighted the need for novel antifungal treatments. Corroborating this finding, drug-resistant fungal isolates have been reported for all known classes of antifungal agents.53 In this context, invasive Pseudallescheria/Scedosporium infections in immunocompromised individuals are characterized by high morbidity and mortality as well as poor response to amphotericin B.54 In their search for possible virulence attributes expressed by this collection of fungi,55 Silva and co-workers56,57 and Pereira and co-workers58 described the production of both secreted and cell-associated metallo-type proteases involved in the cleavage of relevant human protein structures, like serum proteins and extracellular matrix components. As expected, metalloprotease inhibitors (e.g., phen (5) and EDTA) were able to block several essential biological processes in P. boydii, including cell viability, conidia into mycelia differentiation and secretion of proteins.25 These findings suggest that metallo-type enzymes could be potential targets for future therapeutic intervention against P. boydii. The results reported here support this premise, since phen (5) (at the micromolar level) and particularly phendione (6) and its Ag+ and Cu2+ complexes, 12 and 13 (at the nanomolar level) were able to powerfully reduce the conidial viability of P. boydii as compared to the susceptibility of different animal/insect lineages (Table 1). Our observations on the biological effects of metal-free phen (5) and phendione (6) may be attributable to cellular metal chelation.
Larvae of the insect Galleria mellonella (the greater wax moth) were employed to assess the in vivo cytotoxic tolerance of the test compounds (Table 2).32,59,60 Insect larvae have been widely used as a convenient and inexpensive in vivo screening model to assess the therapeutic potential of novel antimicrobial drugs18 and have yielded results that are considered comparable to those obtained using mammalian models.61,62 The innate defences of insects, including G. mellonella, like those of mammals, consist of structural and passive barriers as well as humoral and cellular responses within the haemolymph (analogous to the blood of mammals).60 Indeed, cellular responses within the haemolymph are often activated by signal transduction systems comparable to those in mice, and results obtained using insects strongly correlate with results got from murine testing.63,64 Data for the survival of G. mellonella larvae (expressed as a %) as a function of administered dosages of the test compounds and cisplatin are displayed in Table 2. At the highest administered concentration (100 μg per larvae) 10% of larvae treated with 6 and its Cu(II) and Ag(I) complexes, 12 and 13, respectively, survived, whilst all of the larvae died when injected with 5 and cisplatin. At concentrations of 40 μg per larvae and 20 μg per larvae, 20% of the insects exposed to 5, 6, 12 and 13 survived, whilst all of those subjected to cisplatin perished. At a dosage of 10 μg per larvae all larvae treated with 5, 6, 12 and 13 were alive after 72 h, compared to just 40% survival for those treated with cisplatin. All of the cisplatin-treated larvae survived at a dosage of 2 μg per larvae. Thus, it is clear that cisplatin is considerably more toxic to G. mellonella than 5, 6, 12 and 13. When viewed in terms of complete tolerance (expressed as μmol per larvae) of G. mellonella the relative order is: 5≈6≫13 > 12 > cisplatin.
| Compound | μg per larvae | Administered amount/% mortality | ||||
|---|---|---|---|---|---|---|
| 100 | 40 | 20 | 10 | 2 | ||
| 5 | % Mortality | 100% | 80% | 80% | 0% | 0% |
| μmol | 0.554 | 0.222 | 0.111 | 0.055 | 0.011 | |
| mg kg−1 | 333.3 | 133.3 | 67.67 | 33.33 | 6.67 | |
| 6 | % Mortality | 90% | 80% | 80% | 0% | 0% |
| μmol | 0.475 | 0.190 | 0.095 | 0.047 | 0.001 | |
| mg kg−1 | 333.3 | 133.3 | 67.67 | 33.33 | 6.67 | |
| 12 | % Mortality | 90% | 80% | 80% | 0% | 0% |
| μmol | 0.103 | 0.041 | 0.020 | 0.010 | 0.002 | |
| mg kg−1 | 333.3 | 133.3 | 67.67 | 33.33 | 6.67 | |
| 13 | % Mortality | 90% | 80% | 80% | 0% | 0% |
| μmol | 0.159 | 0.063 | 0.031 | 0.016 | 0.003 | |
| mg kg−1 | 333.3 | 133.3 | 67.67 | 33.33 | 6.67 | |
| Cisplatin | % Mortality | 100% | 100% | 100% | 60% | 0% |
| μmol | 0.333 | 0.133 | 0.066 | 0.033 | 0.007 | |
| mg kg−1 | 333.3 | 133.3 | 67.67 | 33.33 | 6.67 | |
In a related sequence of experiments, Swiss mice were injected with all of the test compounds across the concentration range 15–450 mg kg−1 and mortality (%) monitored every 24, 72 and 168 h (Table 3). Up to a dosage of 45 mg kg−1 no deaths were recorded 168 h post administration of the compounds. When the dose was increased to 150 mg kg−1 all of the mice exposed to 5 and the silver(I) complex, 13, survived. However, at this concentration 20% and 60% of mice treated with 6 and the copper(II) complex, 12, respectively, were dead after 72 h, and the number of deaths increased after a further 96 h (to 80% in each case). A further doubling of the dosage, to 300 mg kg−1, did not induce death in any of the mice exposed to 5. However, at this concentration all of the mice treated with 12 had perished at 24 h and a significant number of those treated with 6 and 13 also died (80% and 40%, respectively) at the same time period. No further fatalities were observed with 6 and 13 after a further 144 h. At the highest dosage used for each compound (450 mg kg−1) there were no survivors at 24 h post administration. At this high concentration, almost immediately after the administration of the compounds, the animals presented strong convulsions, some of them died few hours after injection. In conclusion, the tolerance of Swiss mice is in the order 5 > 13 > 6 > 12. This trend is similar to that observed in the G. mellonella tests, with the exception that the relative tolerance of 6 and 13 was reversed. For G. mellonella and Swiss mice, 5 was the least toxic and the copper(II) complex, 12, was the most.
| Compound | Dose (mg kg−1 d−1) | Dose (mmol kg−1 d−1) | % Mortality | ||
|---|---|---|---|---|---|
| Exposure (h) | |||||
| 24 h | 72 h | 168 h | |||
| 5 | 15 | 0.083 | 0% | 0% | 0% |
| 30 | 0.166 | 0% | 0% | 0% | |
| 45 | 0.249 | 0% | 0% | 0% | |
| 150 | 0.830 | 0% | 0% | 0% | |
| 300 | 1.660 | 0% | 0% | 0% | |
| 450 | 2.790 | 100% | — | — | |
| 6 | 15 | 0.071 | 0% | 0% | 0% |
| 30 | 0.142 | 0% | 0% | 0% | |
| 45 | 0.213 | 0% | 0% | 0% | |
| 150 | 0.710 | 0% | 20% | 80% | |
| 300 | 1.420 | 80% | 80% | 80% | |
| 450 | 2.130 | 100% | — | — | |
| 12 | 15 | 0.016 | 0% | 0% | 0% |
| 30 | 0.032 | 0% | 0% | 0% | |
| 45 | 0.048 | 0% | 0% | 0% | |
| 150 | 0.160 | 0% | 60% | 80% | |
| 300 | 0.320 | 100% | — | — | |
| 450 | 0.480 | 100% | — | — | |
| 13 | 15 | 0.024 | 0% | 0% | 0% |
| 30 | 0.048 | 0% | 0% | 0% | |
| 45 | 0.072 | 0% | 0% | 0% | |
| 150 | 0.240 | 0% | 0% | 0% | |
| 300 | 0.480 | 40% | 40% | 40% | |
| 450 | 0.720 | 100% | — | — | |
Chronic toxicity tests were then conducted in which Swiss mice were inoculated daily with 45 mg kg−1 of each test compound during 5 consecutive days. After this time period, the animals were monitored for an additional 7 days. The results showed that in all cases the mice survived this treatment protocol. Moreover, through visual analyses, the animals remained well throughout the treatment and no behavioural changes were detected. The animals were fed normally and, consequently, there were no differences in body weight of animals treated with PBS (control system), phen (5), phendione (6) or phen-derivatives 12 and 13 (data not shown). At the end of the experiment, the animals were sacrificed and the blood was collected in order to measure two distinct hepatic enzymes, as possible markers for the toxicity of these compounds (Fig. 3). No differences were detected in the level of both hepatic enzymatic activities of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) in non-treated animals and also in those treated with the test compounds.
![Levels of hepatic enzymatic activities (aspartate aminotransferase [AST] and alanine aminotransferase [ALT]) in non-treated Swiss mice and in mice treated with 5, 6, 12 and 13.](/image/article/2012/TX/c2tx00010e/c2tx00010e-f3.gif) | ||
| Fig. 3 Levels of hepatic enzymatic activities (aspartate aminotransferase [AST] and alanine aminotransferase [ALT]) in non-treated Swiss mice and in mice treated with 5, 6, 12 and 13. | ||
Given that the toxicity towards both G. mellonella and Swiss mice of the Ag+ complex, 13, was less than that of the Cu2+ complex, 12, and indeed also that of cisplatin, and also in conjunction with the encouraging AST and ALT enzymatic profile expression, complex 13 was selected for further in vivo investigations. The cytotoxicity of 13 for male nude mice (without tumors) was established by treating the mice with a series of different concentrations of the Ag+ complex (2.5–400 mg kg−1 per inj.) (Table 4). Complex 13 was dissolved in DMSO (end concentration 10%) and further diluted with 0.5% Tween 80 in saline. Mice were injected intraperitoneally (i.p.) with a known concentration of 13 at day 0 only and the effects noted. The complex induced a clear, dose-dependent mortality and body weight loss (BWC). Immediately after injection, the mice showed strong convulsions lasting for about 2 min. At necropsy, inflammation of the gut and pathological livers were obvious (data not shown). The maximum tolerated dose (MTD) of 13 was estimated to be 10 mg kg−1 (0.016 mmol kg−1). It is interesting to note that Swiss mice appear to withstand up to 150 mg kg−1 of 13 in the acute toxicity tests (and 45 mg kg−1 in chronic toxicity tests), whereas the nude mice (without implanted tumors) can only tolerate 10 mg kg−1 of the Ag+ complex.
| Group | Nude mice | Tested | Treatment | Dose | Toxic | BWCb |
|---|---|---|---|---|---|---|
| Tested | Compound | (d)a | (mg kg−1 per inj.) | Deaths (d)a | (%) | |
| a d = day. b BWC = body weight change. | ||||||
| 1A | 2 | 13 | 0 | 2.5 | 0 | 0 |
| 1B | 2 | 13 | 0 | 5 | 0 | −2 |
| 1C | 2 | 13 | 0 | 10 | 0 | −11 |
| 1D | 2 | 13 | 0 | 20 | 2 (5) | −20 |
| 2A | 2 | 13 | 0 | 25 | 2 (3) | −9 |
| 2B | 2 | 13 | 0 | 50 | 2 (3) | −7 |
| 2C | 2 | 13 | 0 | 100 | 2 (1) | n.t. |
| 2D | 2 | 13 | 0 | 200 | 2 (1) | n.t. |
| 2E | 2 | 13 | 0 | 400 | 2 (1) | n.t. |
A similar xenograft study was conducted using solutions of 13 on nude mice transplanted with human HCT-8 colon carcinoma cells. In total, 24 mice were transplanted with the HCT-8 cells and all were included in the treatment and evaluation. Treatment started at palpable tumors (day 8). In two groups, 13 was again administered at doses of 5 and 10 mg kg−1 per injection on days 8, 12 and 16 (Table 5 and Fig. 4). The experiment was terminated at day 22 because of the appearance of large tumors in some mice. Again, 13 had no influence on tumor growth at either of the administered doses. As was the case with the Hep-G2 xenografts, 13 induced a dose-dependent body weight loss, but had no influence on blood parameter.
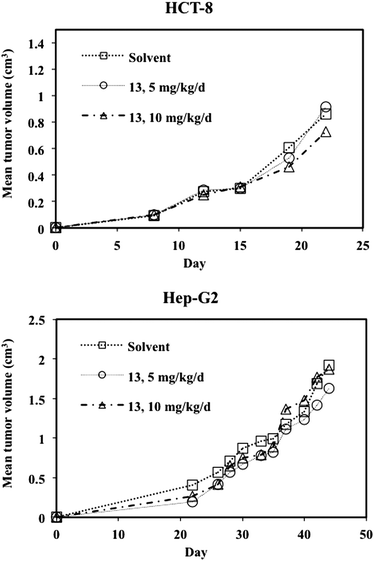 | ||
| Fig. 4 Development of tumor volume for nude mice transplanted with human Hep-G2 and HCT-8 cells. | ||
| Group | Nude mice | Tested | Treatment | Dose | Toxic | BWCb | Optimum | WBCc | Thromb.e |
|---|---|---|---|---|---|---|---|---|---|
| Tested | Substance | (d)a | (mg kg−1 per inj.) | Deaths (d)a | % | T/Cd % | (106 mL−1) | (106 mL−1) | |
| a d = day. b BWC = body weight change. c WBC = white blood cells. d T/C = treated to control. e thromb. = thrombocytes. | |||||||||
| Hep-G2 (hepatocellular carcinoma) 107 cells s.c. (day 0) | |||||||||
| d 22–33 | d 26 | d 26 | |||||||
| A | 5 | Solvent | 22, 26, 30 | 0 | — | 0 | — | 9.6 ± 2.9 | 1204 ± 72 |
| B | 5 | 13 | 22, 26, 30 | 5 | 0 | −2 | 91 | 9.6 ± 2.3 | 1322 ± 120 |
| C | 5 | 13 | 22, 26, 30 | 10 | 0 | −11 | 87 | 12.6 ± 2 | 1428 ± 66 |
| HCT-8 (colon carcinoma), 107 cells s.c. (day 0) | |||||||||
| d 8–22 | d 12 | d 12 | |||||||
| A | 8 | Solvent | 8, 12, 16 | 0 | — | −7 | — | 6.9 ± 0.6 | 1289 ± 118 |
| B | 8 | 13 | 8, 12, 16 | 5 | 0 | −4 | 75 | 10.2 ± 1 | 1235 ± 99 |
| C | 8 | 13 | 8, 12, 16 | 10 | 0 | −14 | 71 | 10.2± | 1122 ± 276 |
The results obtained here for complex 13 contrast with those previously reported for the treatment of implanted human-derived solid tumors by the V(IV) dimethylated-phen complex, METVAN (bis(4,7-dimethyl-1,10-phenanthroline)-sulfato-oxovanadium(IV) {[VO(SO4)(Me2-Phen)2]}),1 and the La(III) complex, KP772 {tris(1,10-phenanthroline)-lanthanum(III)-trithiocyanate}.8 METVAN, administered (i.p.) for 5 days per week for 4 weeks at 10 mg kg−1 d−1, resulted in significant delayed tumor progression in SCID mouse xenograft models against both human glioblastoma and breast cancer. Additionally, KP772, administered (intravenously) on days 0–4 at 4, 8 or 12 mg kg−1 d−1 also displayed significant, dose-dependent, delayed tumor progression toward DLD-1 human colon implanted mice by day 14. These results compared favorably to the parallel xenograft experiment conducted on the reduction in tumor volume by cisplatin, which was administered at 2 mg kg−1 d−1 between days 0–4.8 However, it should be highlighted that in Rosenberg et al.'s original report on the in vivo efficacy of cisplatin against implanted sarcoma-180, administered by single injection at 8 mg kg−1 on day 8, complete eradication of that particular tumor was observed by day 36.65 It should be noted however, direct comparisons between the intravenously applied agent KP772 and 13, which was applied intraperitoneally, are not possible here, particularly as the interaction with serum iron could be significant in the anticancer activity of phenanthroline compounds.
Conclusions
Derivatization of the ligand phen (5) to phen-dione (6) results in a substantial cytotoxic enhancement against nearly all cell lineages tested. Moreover, upon complexation of 6 to yield [Cu(phendione)3](ClO4)2·4H2O (12) and [Ag(phendione)2]ClO4 (13) a further enhancement is evident, particularly against the human-derived tumor lines (MRC-5, HEp-2, A549, DLKP, A498 and Hep-G2) with most IC50 (μM) values decreasing by at least 50% compared with the metal-free phendione (6) ligand. Cytoselectivity was not observed as both metal complexes were more toxic toward the non-cancerous liver (Chang) and renal (HK-2) cells compared to the respective tumor lines, Hep-G2 and A498. While the Cu2+ complex, 12, was more active against the multi-resistant fungi P. boydii, than its Ag+ counterpart, 13, it was the latter complex which exhibited better activity against C. albicans. Overall, both 12 and 13 can be described as potent cytotoxic agents capable of eliciting low-micromolar or nanomolar cytotoxicities against both fungal and human-derived cell lines.Against the insect larvae, G. mellonella, the metal-free ligands 5 and 6 displayed the greatest tolerance in vivo and complexes 12 and 13 were significantly less toxic than cisplatin. Encouragingly, at a concentration of 33.3 μg kg−1 the mortality induced by 12 and 13 was 0%, compared to 60% for cisplatin at the same concentration. For Swiss mice, the tolerance trend was 5 > 13 > 6 > 12, and at an administered dose of 150 mg kg−1 d−1, the Ag+ complex, 12, did not cause fatalities, whilst with the metal-free phendione ligand (6) there was 80% mortality over the same 7 day period. In chronic in vivo toxicity studies, using Swiss mice exposed to 45 mg kg−1 d−1 of 5, 6, 12 and 13 over 1 week, the animals remained well throughout and there were no obvious behavioural changes and no detectable differences in the expression levels of AST and ALT hepatic enzymes. It is evident that the compounds are well tolerated by both G. mellonella and Swiss mice. In contrast to the relatively high tolerance of Swiss mice to [Ag(phendione)2]ClO4 (13) the MTD of immunocompromised nude mice was significantly lower (10 mg kg−1 d−1). Higher concentrations of 13 results in mortality coupled with unacceptable % body weight changes in the animals. Administering 13 (at a concentration of either 5 or 10 mg kg−1 d−1) to nude mice implanted with Hep-G2 (hepatic) or HCT-8 (colon) human-derived tumors had no influence on tumor growth. Thus, in their current form, these Cu(II) and Ag(I) phendione compounds appear to offer excellent potential for the cytotoxic treatment of both fungi and insects, however, structural modifications will be required for the translation of this exceptional in vitro cytotoxicity into an acceptable in vivo antitumor effect.
Acknowledgements
This study was supported by grants from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ). M. McCann, A. Kellett and M. Devereux would like to thank Prof. R. O'Neill from the office of Vice President for Research, NUI Maynooth, and the Dublin Institute of Technology Capacity Building Scheme for Strategic Research (CaBS) for generous assistance with the cost of the xenograft studies.Notes and references
- R. K. Narla, C. L. Chen, Y. Dong and F. M. Uckun, Clin. Cancer. Res., 2001, 7, 2124–2133 CAS.
- R. K. Narla, Y. Dong, D. Klis and F. M. Uckun, Clin. Cancer. Res., 2001, 7, 1094–1101 CAS.
- S. Tardito and L. Marchio, Curr. Med. Chem., 2009, 16, 1325–1348 CrossRef CAS.
- L. Ruiz-Azuara and M. E. Bravo-Gomez, Curr. Med. Chem., 2010, 17, 3606–3615 CrossRef CAS.
- C. Deegan, M. McCann, M. Devereux, B. Coyle and D. A. Egan, Cancer Lett., 2007, 247, 224–233 CrossRef CAS.
- S. Roy, K. D. Hagen, P. U. Maheswari, M. Lutz, A. L. Spek, J. Reedijk and G. P. van Wezel, ChemMedChem, 2008, 3, 1427–1434 CrossRef CAS.
- D. Igdaloff, D. V. Santi, T. S. Eckert and T. C. Bruice, Biochem. Pharmacol., 1983, 32, 172–174 CrossRef CAS.
- P. Heffeter, M. A. Jakupec, W. Korner, S. Wild, N. G. von Keyserlingk, L. Elbling, H. Zorbas, A. Korynevska, S. Knasmuller, H. Sutterluty, M. Micksche, B. K. Keppler and W. Berger, Biochem. Pharmacol., 2006, 71, 426–440 CrossRef CAS.
- A. D. Randford and P. J. Sadler, J. Chem. Soc., Dalton Trans., 1993, 3393–3399 RSC.
- N. Margiotta, A. Bergamo, G. Sava, G. Padovano, E. de Clercq and G. Natile, J. Inorg. Biochem., 2004, 98, 1385–1390 CrossRef CAS.
- P. Papadia, N. Margiotta, A. Bergamo, G. Sava and G. Natile, J. Med. Chem., 2005, 48, 3364–3371 CrossRef CAS.
- R. A. Macleod, J. Biol. Chem., 1952, 197, 751–761 CAS.
- R. Husseini and R. J. Stretton, Microbios, 1980, 29, 109–125 CAS.
- R. Husseini and R. J. Stretton, Microbios, 1981, 30, 7–18 CAS.
- H. S. Husseini and R. J. Stretton, Microbios. Lett., 1981, 16, 85–94 Search PubMed.
- M. McCann, M. Geraghty, M. Devereux, D. O'Shea, J. Mason and L. O'Sullivan, Met.-Based Drugs, 2000, 7, 185–193 CrossRef CAS.
- B. Coyle, K. Kavanagh, M. McCann, M. Devereux and M. Geraghty, BioMetals, 2003, 16, 321–329 CrossRef CAS.
- R. Rowan, C. Moran, M. McCann and K. Kavanagh, BioMetals, 2009, 22, 461–467 CrossRef CAS.
- B. C. Baguley, W. A. Denny, G. J. Atwell and B. F. Cain, J. Med. Chem., 1981, 24, 520–525 CrossRef CAS.
- B. C. Baguley, W. A. Denny, G. J. Atwell and B. F. Cain, J. Med. Chem., 1981, 24, 170–177 CrossRef CAS.
- B. D. Palmer, H. H. Lee, P. Johnson, B. C. Baguley, G. Wickham, L. P. Wakelin, W. D. McFadyen and W. A. Denny, J. Med. Chem., 1990, 33, 3008–3014 CrossRef CAS.
- P. Cudic, M. Zinic, V. Tomisic, V. Simeon, J.-P. Vigneron and J.-M. Lehn, J. Chem. Soc., Chem. Commun., 1995, 1073–1075 RSC.
- M. McCann, B. Coyle, S. McKay, P. McCormack, K. Kavanagh, M. Devereux, V. McKee, P. Kinsella, R. O'Connor and M. Clynes, BioMetals, 2004, 17, 635–645 CrossRef CAS.
- C. Deegan, B. Coyle, M. McCann, M. Devereux and D. A. Egan, Chem.-Biol. Interact., 2006, 164, 115–125 CrossRef CAS.
- B. A. Silva, A. L. Souza-Goncalves, M. R. Pinto, E. Barreto-Bergter and A. L. Santos, Mycoses, 2011, 54, 105–112 CrossRef CAS.
- B. M. Zeglis, V. C. Pierre and J. K. Barton, Chem. Commun., 2007, 4565–4579 RSC.
- H. K. Liu and P. J. Sadler, Acc. Chem. Res., 2011 Search PubMed.
- M. Pitie and G. Pratviel, Chem. Rev., 2010, 110, 1018–1059 CrossRef CAS.
- D. S. Sigman, D. R. Graham, V. D'Aurora and A. M. Stern, J. Biol. Chem., 1979, 254, 12269–12272 CAS.
- S. N. Georgiades, N. H. AbdKarim, K. Suntharalingam and R. Vilar, Angew. Chem., Int. Ed., 2010, 49, 4020–4034 CrossRef CAS.
- J. Talib, C. Green, K. J. Davis, T. Urathamakul, J. L. Beck, J. R. Aldrich-Wright and S. F. Ralph, Dalton Trans., 2008, 1018–1026 RSC.
- A. Kellett, M. O'Connor, M. McCann, O. Howe, A. Casey, P. McCarron, K. Kavanagh, M. McNamara, S. Kennedy, D. D. May, P. S. Skell, D. O'Shea and M. Devereux, Med. Chem. Commun., 2011, 2, 579–676 RSC.
- C. Marzano, M. Pellei, F. Tisato and C. Santini, Anti-cancer Agents Med. Chem., 2009, 9, 185–211 CAS.
- N. Marino, A. R. Vortherms, A. E. Hoffman and R. P. Doyle, Inorg. Chem., 2010, 49, 6790–6792 CrossRef CAS.
- O. F. Ikotun, E. M. Higbee, W. Ouellette and R. P. Doyle, J. Inorg. Biochem., 2009, 103, 1254–1264 CrossRef CAS.
- E. Ueta, T. Tanida, K. Yoneda, T. Yamamoto and T. Osaki, Oral Microbiol. Immunol., 2001, 16, 243–249 CrossRef CAS.
- Y. Sun, J. Bian, Y. Wang and C. Jacobs, Oncogene, 1997, 14, 385–393 CAS.
- V. W.-W. Yam, K. K.-W. Lo, K.-K. Cheung and R. Y.-C. Kong, J. Chem. Soc., Chem. Commun., 1995, 1191–1193 RSC.
- P. K. Sasmal, S. Saha, R. Majumdar, R. R. Dighe and A. R. Chakravarty, Inorg. Chem., 2010, 49, 849–859 CrossRef CAS.
- S. Dhar, M. Nethaji and A. R. Chakravarty, Inorg. Chem., 2005, 44, 8876–8883 CrossRef CAS.
- P. K. Sasmal, S. Saha, R. Majumdar, S. De, R. R. Dighe and A. R. Chakravarty, Dalton Trans., 2010, 39, 2147–2158 RSC.
- T. N. Singh and C. Turro, Inorg. Chem., 2004, 43, 7260–7262 CrossRef CAS.
- E. L. Menon, R. Perera, M. Navarro, R. J. Kuhn and H. Morrison, Inorg. Chem., 2004, 43, 5373–5381 CrossRef CAS.
- M. Devereux, D. O Shea, A. Kellett, M. McCann, M. Walsh, D. Egan, C. Deegan, K. Kedziora, G. Rosair and H. Müller-Bunz, J. Inorg. Biochem., 2007, 101, 881–892 CrossRef CAS.
- M. Devereux, M. McCann, D. O Shea, M. O'Connor, E. Kiely, V. McKee, D. Naughton, A. Fisher, A. Kellett, M. Walsh, D. Egan and C. Deegan, Bioinorg. Chem. Appl., 2006, 2006 Search PubMed.
- M. Geraghty, V. Sheridan, M. McCann, M. Devereux and V. McKee, Polyhedron, 1999, 18, 2931–2939 CrossRef CAS.
- A. Eshwika, B. Coyle, M. Devereux, M. McCann and K. Kavanagh, BioMetals, 2004, 17, 415–422 CrossRef CAS.
- A. Kellett, M. O'Connor, M. McCann, M. McNamara, P. Lynch, G. Rosair, V. McKee, B. Creaven, M. Walsh, S. McClean, A. Foltyn, D. O'Shea, O. Howe and M. Devereux, Dalton Trans., 2011, 40, 1024–1027 RSC.
- A. D. Khalaji, A. M. Slawin and J. D. Woolins, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m2848 Search PubMed.
- M. Yamada, Y. Tanaka, Y. Yoshimoto, S. Kurodo and I. Shimao, Bull. Chem. Soc. Jpn., 1992, 65, 1006 CrossRef CAS.
- F. Gilgado, J. Cano, J. Gene and J. Guarro, J. Clin. Microbiol., 2005, 43, 4930–4942 CrossRef CAS.
- E. Borenfreund and J. A. Puerner, Toxicol. Lett., 1985, 24, 119–124 CrossRef CAS.
- C. C. Lai, C. K. Tan, Y. T. Huang, P. L. Shao and P. R. Hsueh, J. Infect. Chemother., 2008, 14, 77–85 CrossRef CAS.
- K. J. Cortez, E. Roilides, F. Quiroz-Telles, J. Meletiadis, C. Antachopoulos, T. Knudsen, W. Buchanan, J. Milanovich, D. A. Sutton, A. Fothergill, M. G. Rinaldi, Y. R. Shea, T. Zaoutis, S. Kottilil and T. J. Walsh, Clin. Microbiol. Rev., 2008, 21, 157–197 CrossRef CAS.
- A. L. Santos, V. C. Bittencourt, M. R. Pinto, B. A. Silva and E. Barreto-Bergter, Med. Mycol., 2009, 47, 375–386 CrossRef CAS.
- B. A. da Silva, A. L. dos Santos, E. Barreto-Bergter and M. R. Pinto, Curr. Microbiol., 2006, 53, 18–22 CrossRef.
- B. A. Silva, M. R. Pinto, R. M. Soares, E. Barreto-Bergter and A. L. Santos, Res. Microbiol., 2006, 157, 425–432 CrossRef CAS.
- M. M. Pereira, B. A. Silva, M. R. Pinto, E. Barreto-Bergter and A. L. dos Santos, Mycopathologia, 2009, 167, 25–30 CrossRef CAS.
- K. Kavanagh and J. P. Fallon, Fungal Biol. Rev., 24, 79–83 Search PubMed.
- K. Kavanagh and E. P. Reeves, FEMS Microbiol. Rev., 2004, 28, 101–112 CrossRef CAS.
- H. Hamamoto, K. Kurokawa, C. Kaito, K. Kamura, I. Manitra Razanajatovo, H. Kusuhara, T. Santa and K. Sekimizu, Antimicrob. Agents Chemother., 2004, 48, 774–779 CrossRef CAS.
- H. Hamamoto, A. Tonoike, K. Narushima, R. Horie and K. Sekimizu, Comp. Biochem. Physiol., Part C: Toxicol. Pharmacol., 2009, 149, 334–339 CrossRef.
- G. Jander, L. G. Rahme and F. M. Ausubel, J. Bacteriol., 2000, 182, 3843–3845 CrossRef CAS.
- M. Brennan, D. Y. Thomas, M. Whiteway and K. Kavanagh, FEMS Immunol. Med. Microbiol., 2002, 34, 153–157 CrossRef CAS.
- B. Rosenberg, L. VanCamp, J. E. Trosko and V. H. Mansour, Nature, 1969, 222, 385–386 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |