Mitochondrial glutathione in toxicology and disease of the kidneys
Lawrence H.
Lash
*
Department of Pharmacology, Wayne State University School of Medicine, 540 East Canfield Avenue, Detroit, MI 48201, USA. E-mail: l.h.lash@wayne.edu; Fax: +01-313-577-6739; Tel: +01-313-577-0475
First published on 9th May 2012
Abstract
The tripeptide glutathione (GSH), comprised of the amino acids L-cysteine, glycine, and L-glutamate, is found in all cells of aerobic organisms and plays numerous, critical roles as an antioxidant and nucleophile in regulating cellular homeostasis and drug metabolism. GSH is synthesized exclusively in the cytoplasm of most cells by two ATP-dependent reactions. Despite this compartmentation, GSH is found in other subcellular compartments, including mitochondria. As the GSH molecule has a net negative charge at physiological pH, it cannot cross cellular membranes by diffusion. Rather, GSH is a substrate for a variety of anion and amino acid transporters. Two organic anion carriers in the inner membrane of renal mitochondria, the dicarboxylate carrier (DIC; Slc25a10) and the 2-oxoglutarate carrier (OGC; Slc25a11), are responsible for most of the transport of GSH from cytoplasm into mitochondrial matrix. Genetic manipulation of DIC and/or OGC expression in renal cell lines demonstrated the ability to produce sustained increases in mitochondrial GSH content, which then protected these cells from cytotoxicity due to several oxidants and mitochondrial toxicants. Several diseases and pathological states are associated with mitochondrial dysfunction and oxidative stress, suggesting that the mitochondrial GSH pool may be a therapeutic target. One such disease that is of particular concern for public health is diabetic nephropathy. Another chronic, pathological state that is associated with bioenergetic and redox changes is compensatory renal hypertrophy that results from reductions in functional renal mass. This review summarizes pathways of mitochondrial GSH transport and discusses studies on its manipulation in toxicological and pathological states.
 Lawrence H. Lash | Lawrence H. Lash, Ph.D. is a Professor and Associate Chair in the Department of Pharmacology at Wayne State University School of Medicine in Detroit, Michigan. Dr. Lash received his Ph.D. in Biochemistry from Emory University (1985) with Dean Jones and was a postdoctoral fellow with M. W. Anders at the University of Rochester (1985–1988). Dr. Lash's research focuses on renal drug metabolism and toxicology. His laboratory identified the carriers responsible for transport of glutathione into renal mitochondria and established in vitro cell models using rat and human kidney for use in metabolic, dispositional and toxicological studies of various renal toxicants. |
Biochemistry of glutathione (GSH) in the kidneys
GSH is a tripeptide composed of the amino acids L-glutamate, L-cysteine, and glycine (Fig. 1). Due to the low pKa of the two free carboxyl groups, the GSH molecule has a net negative charge at physiological pH. The pKa of the sulfhydryl or thiol (SH) group is only 8.3, indicating that a significant proportion of the SH groups will be deprotonated at slightly alkaline pH values, such as those typically found in the mitochondrial matrix (pH = ∼7.8). It is present at millimolar concentrations in virtually all cells of aerobic organisms. The GSH molecule has two unique features that are responsible for its myriad of functions. The first feature is the SH group on the cysteinyl residue. The SH group provides GSH with all its functions, which can be divided into actions as a reductant and a nucleophile. GSH is thus critical for the maintenance of redox homeostasis, numerous drug metabolism reactions, and for processes that are mechanistically linked to redox status, such as opening and closing of the mitochondrial permeability transition pore and the mitochondrial pathway of apoptosis.1–7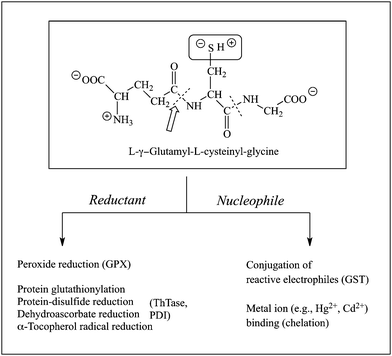 | ||
| Fig. 1 Structure and major functions of GSH. Abbreviations: GPX, GSH peroxidase; GST, GSH S-transferase; PDI, protein-disulfide isomerase; ThTase, thioltransferase. | ||
The second feature is the isopeptide linkage between the L-glutamyl and L-cysteinyl residues, which makes the GSH molecule resistant to most proteases. As a consequence of this linkage between the γ-carboxyl group of the L-glutamyl residue and the α-amino group of the L-cysteinyl residue, the GSH molecule can be transported out of tissues and translocated through the plasma without being degraded.
GSH synthesis occurs in the cytoplasm of most cells and is catalysed by two ATP-dependent enzymes, the glutamate-cysteine ligase (GCL; also called γ-glutamylcysteine synthetase; EC 6.3.2.2) [Reaction 1], which forms L-γ-glutamyl-L-cysteine, and GSH synthetase (GS; EC 6.3.2.3) [Reaction 2], which forms GSH (Fig. 2). GSH undergoes turnover by cleavage of the isopeptide or γ-glutamyl peptide bond, as catalysed by γ-glutamyltransferase (GGT; EC 2.3.2.2) [Reaction 3], which releases L-glutamate (when GGT functions as a hydrolase) or a γ-glutamyl amino acid or peptide when GGT functions as a transpeptidase and uses a γ-glutamyl acceptor instead of water) and L-cysteinylglycine. The latter dipeptide is then hydrolysed by a dipeptidase to release L-cysteine and glycine [Reaction 4]. By having distinct synthetic and degradative pathways, intracellular concentrations of GSH can be better regulated.
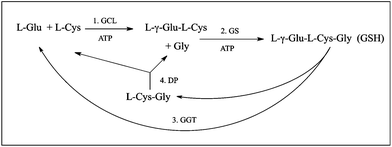 | ||
| Fig. 2 Pathways for synthesis and degradation of GSH. Abbreviations: DP, dipeptidase; GCL, glutamate-cysteine ligase; GGT, γ-glutamyltransferase; GS, GSH synthetase. | ||
Although GSH synthesis occurs in most tissues, albeit at different rates, the enzymes of the degradation pathway are not uniformly distributed. Rather, GSH-degrading capacity, which is primarily controlled by GGT activity, is highest in the kidneys and is also present at significant levels in the small-intestinal epithelium and choroid plexus.8 In fact, one of the unique features of renal handling of GSH is the extremely high activity of GGT, which is localized on the brush-border or luminal membrane of the proximal tubular (PT) cell. GGT is an integral membrane protein whose active site faces the exterior of the PT cell. This is critically important for GSH homeostasis because it prevents intracellular GSH from being degraded without it being transported out of the cell into the tubular lumen.
The other unique feature of renal handling of GSH is that the kidneys extract a majority (i.e., 80%) of plasma GSH that passes through the renal circulation; this, coupled with the extremely high activity of GGT on the brush-border plasma membrane of renal PT cells, makes the kidneys the primary tissue for turnover of GSH in the body.9 Renal extraction of plasma GSH occurs by two mechanisms, glomerular filtration and active basolateral uptake from the renal periplasmic space. As the glomerulus only filters ∼30% of plasma during a single pass of the blood through the kidneys, this indicates that the basolateral transport mechanism accounts for nearly two-thirds of the total extraction by the kidneys. As noted above, intracellular GSH must be transported across the brush-border plasma membrane out of the PT cell to be degraded by GGT. Thus, it is clear that membrane transport processes play a central role in the renal handling of GSH. This prominent role of transport is relatively unique for the kidneys because most tissues do not transport intact GSH into the cell, although efflux is a relatively common process.
Roles of GSH in mitochondrial function
Although the major intracellular pool of GSH is in the cytoplasm, other subcellular organelles contain separate pools of GSH. In particular, renal and hepatic mitochondria have been shown to contain a separate pool of GSH that has a much slower turnover and distinct mechanism of regulation as compared to the major intracellular pool in the cytoplasm.10–12 Moreover, as noted above, while GSH synthesis occurs in virtually all cells, the two ATP-dependent enzymes that catalyze GSH synthesis appear to be exclusively localized in the cytoplasm.13,14 Hence, the mitochondrial pool of GSH must derive from the cytoplasm by a mechanism involving carrier-mediated transport. While the primary focus of this review is to summarize the importance of mitochondrial GSH in disease states and chemical toxicant exposures and how manipulation of the transport process can alter mitochondrial and renal cellular function, it is first important to consider some key aspects of how GSH functions in regulating basic mitochondrial function.One of the most critical functions of the mitochondrial GSH pool can be surmised from a consideration of the abundance of enzymes and transporters that possess critical cysteinyl residues that must be maintained in the reduced form to retain maximal activity.15 Among these proteins are the F0F1-ATPase, numerous dehydrogenases, and several of the inner membrane carrier proteins. Additionally, there are redox-sensitive components in the electron transport chain, such as iron ions, iron–sulfur centers, and heme prosthetic groups of the various cytochromes that are subject to oxidative modification and functional alteration. The importance of having an adequate supply of GSH in the mitochondria is further highlighted by the fact that mitochondria are the primary intracellular sites of oxygen consumption in most cells and may also be the primary sites for generation of reactive oxygen species (ROS) during pathological or toxicological states. Maintenance of proper concentrations and redox state of the mitochondrial GSH pool is critical for processes such as Ca2+ ion distribution, pyridine nucleotide redox state, protection of mitochondrial DNA from damage, and appropriate regulation of the mitochondrial membrane permeability transition, as reviewed previously.16 Besides a role in regulation of mitochondrial responses to ROS, GSH can also interact with reactive nitrogen species (RNS), such as nitric oxide (NO) and peroxynitrite (ONOO−). As summarized previously,16 GSH may react with NO to form S-nitrosoglutathione, which may serve as an NO donor or may promote nitrosylation of proteins, leading to alterations in their function.
Pathways of GSH transport into renal mitochondria
Although there has been interest in mitochondrial GSH since the 1960s17–21 and identification of a distinctly regulated mitochondrial pool was first reported in the early 1980s,10 the concept that GSH was transported into mitochondria was not considered until about 1990.22,23 A few other studies in that era24,25 demonstrated different activities of mitochondrial GSH transport under different redox or energetic conditions. To better understand the mechanism of GSH transport into mitochondria and to investigate its potential utility as a therapeutic target against the large number of pathological and toxicological states that are associated with disturbances in the mitochondrial GSH pool, however, it was necessary to identify the specific carrier proteins responsible for the transport.Identification of potential carriers for GSH in the inner mitochondrial membrane was first suggested by studies of Lash and colleagues.14 Suggestion of the potential function of two specific carriers in the mitochondrial inner membrane, the dicarboxylate carrier (DIC; Slc25a10) and 2-oxoglutarate carrier (OGC; Slc25a11), in GSH transport (Fig. 3) was based on several considerations: (1) GSH is an anion at physiological pH, (2) a membrane potential and pH gradient exist across the mitochondrial inner membrane, (3) mitochondria possess a large family of carriers (the Slc25 gene family) that transport small organic anions, such as certain amino acids and citric acid cycle intermediates,26–28 and (4) the rather broad substrate specificity typical of these carriers. Further studies by Lash and colleagues,29,30 involving analysis of substrate specificity and transport energetics, validated the predominant function of these two inner membrane carrier proteins in the transport of GSH into renal cortical mitochondria. It is important to note that at physiological pH, the GSH molecule (shown as GS− in Fig. 3) possesses two negative charges from two free carboxyl groups and a positive charge from a free amino group. Moreover, a significant proportion of the –SH group may be deprotonated because of its fairly low pKa (= 8.3), making the GSH molecule have a net charge of −2. Most of the molecules, however, will have a net charge of −1.
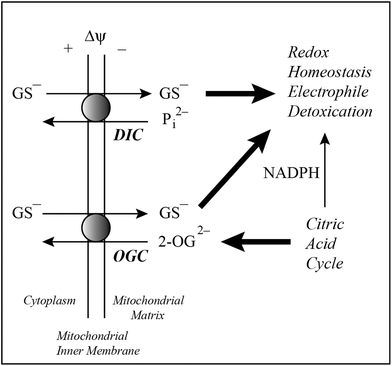 | ||
| Fig. 3 Pathways of mitochondrial GSH transport. Two carriers on the mitochondrial inner membrane of renal cortex, the dicarboxylate carrier (DIC; Slc25a10) and 2-oxoglutarate carrier (OGC; Slc25a11), are responsible for most of the observable transport of GSH from cytoplasm into mitochondrial matrix. The DIC can exchange GSH for inorganic phosphate (Pi2−) whereas the OGC exchanges GSH for 2-oxoglutarate (2-OG). Both transport processes integrate with and are critical for the citric acid cycle, pyridine nucleotides, and maintenance of redox homeostasis. | ||
Although the focus of this review is on the importance of mitochondrial GSH in renal toxicology and disease, some brief discussion is warranted about the general applicability of these transport processes to mitochondria in other tissues. The DIC and OGC are present in all mitochondria, although tissue-specific differences in abundance do exist.16,27 With regard to GSH transport, function of the DIC and/or OGC have been documented in mitochondria of other tissues, including those of the liver,31,32 brain,33,34 and colonic epithelial cells.35 Besides differences in abundance, additional carriers may function in GSH transport, such as the oxodicarboxylate carrier in liver mitochondria.32 Nonetheless, despite some tissue-dependent differences, the DIC and OGC play the predominant roles in mitochondrial GSH transport in renal epithelial cells and elsewhere.
Manipulation of mitochondrial GSH transporter expression alters susceptibility of NRK-52E cells to chemically induced injury
Identification of the DIC and OGC as the principal inner membrane carriers responsible for uptake of cytoplasmic GSH, which is the process that determines mitochondrial matrix GSH content,16 opens up a new avenue for potential therapeutic approaches. Cellular and subcellular GSH concentrations are tightly regulated by a variety of mechanisms, including feedback inhibition of GCL. Administration of either GSH itself, GSH esters, or precursors that result in increased intracellular synthesis of GSH, can produce increases in intracellular GSH content during conditions in which the GSH pool is depleted or oxidized. These increases, however, are typically transient and GSH levels return to their previous set point. To produce a more permanent increase in GSH levels, to combat decreases or oxidation due to certain diseases or exposures to drugs or environmental contaminants, other approaches are needed. To address this need, Lash and colleagues hypothesized that overexpression of the cDNAs for either the DIC36 or OGC37 could rescue renal proximal tubular cells from cytotoxicity induced by various oxidants or mitochondrial toxicants.NRK-52E cells, which are a commercially available immortalized cell line derived from Normal Rat Kidney Epithelial cells of proximal tubular origin, were used as a model system to investigate the physiological functions and toxicological implications of genetic manipulation of mitochondrial GSH transporters.36–38 These cells have reasonably high content of mitochondria, express many properties of renal proximal tubular cells, but have low activities of several GSH transport carriers.38 These characteristics make the NRK-52E cells good models for the purpose of manipulating mitochondrial GSH transporter expression because the relatively low background activity simplifies interpretation of results.
To probe the toxicological effects of modulation of DIC or OGC expression, NRK-52E cells were either transiently or stably transfected with cDNAs for either the DIC or OGC, respectively. Transport of GSH (Fig. 4A) was markedly increased in mitochondria of transfected cells. In particular, rates of GSH uptake in mitochondria of NRK-52E cells stably transfected with OGC cDNA were nearly 20-fold higher than those of mitochondria from non-transfected cells (designated as wild-type; WT), using either GSH or 2-OG as substrate. Additionally, a double-cysteine mutant of the OGC in which two L-cysteinyl residues that normally form an intramolecular disulfide bond were converted to L-serine (C221,224S), was generated by site-directed mutagenesis and exhibited <25% of the transport activity of the normal carrier. Additionally, mitochondria from cells stably transfected with the C221,224S mutant exhibited GSH concentrations that were only 40% to 80% of those from non-transfected cells.
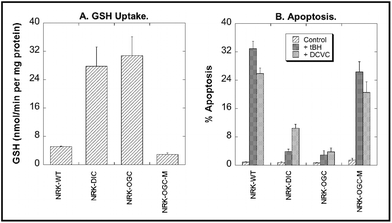 | ||
| Fig. 4 Modulation of DIC or OGC expression in NRK-52E cells alters mitochondrial GSH uptake and susceptibility to chemically induced apoptosis. NRK-52E cells were transfected using FuGENE 6 with cDNA for either the rat DIC (transient), rat OGC (stable), or C221,224S mutant OGC (OGC-M) using the pcDNA3.1/V5-His-TOPO vector. Non-transfected cells are designed as wild-type (WT). Panel A: Initial rates of GSH uptake were measured using radiolabeled (3H-glycyl) GSH. Panel B: Cells were incubated for 4 h with either culture medium (= Control), 50 μM tert-butyl hydroperoxide (tBH), or 200 μM S-(1,2-dichlorovinyl)-L-cysteine (DCVC). Apoptosis was quantified by propidium iodide staining, flow cytometry, and flow-activated cell sorting (FACS) analysis. (Data are a compilation from multiple studies, so statistical comparisons could not be performed. See ref. 36 and 37 for more Experimental details.) | ||
To explore the toxicological relevance of modulation of mitochondrial GSH transport activity with either of the two carriers or the OGC double-cysteine mutant, WT and transfected NRK-52E cells were incubated for 4 h with either cell culture medium (= Control), 50 μM tert-butyl hydroperoxide (tBH), or 200 μM S-(1,2-dichlorovinyl)-L-cysteine (DCVC). Induction of apoptosis was then assessed by flow cytometry and cell cycle analysis after staining of cells with propidium iodide (Fig. 4B). tBH is a potent mitochondrial nephrotoxicant that acts by causing GSH oxidation and lipid peroxidation.14 In contrast, DCVC, which is the penultimate nephrotoxic metabolite of the environmental contaminant trichloroethylene, causes mitochondrial dysfunction as an early step,39,40 with the underlying mechanism involving both GSH oxidation and thiol or protein sulfhydryl group alkylation.41 Both tBH and DCVC caused a large increase in apoptosis in NRK-52E-WT cells. Overexpression of either carrier markedly decreased the fraction of cells undergoing apoptosis whereas overexpression of the OGC double-cysteine mutant had little effect on tBH- or DCVC-induced apoptosis.
These results validate the hypothesis that overexpression of either of the two mitochondrial GSH transporters in renal cells results in sustained increases in mitochondrial GSH content and decreased susceptibility to oxidative injury. Furthermore, the lack of protection when a mutant OGC protein was overexpressed, suggests that a threshold concentration of GSH exists below which renal proximal tubular cells are highly sensitive to mitochondrial toxicants or other oxidants.
Mitochondrial GSH in diabetic nephropathy
Although demonstration of toxicological relevance is important for understanding drug-induced nephrotoxicity, potential roles of mitochondrial GSH in either the etiology of human disease or development of therapeutic approaches to treat human disease are of greatest interest in this era of translational research. The increasing incidence of diabetes in Western society highlights the need to develop novel therapeutic approaches to treat both the underlying disease as well as the various complications that arise. One of the major complications of diabetes is nephropathy, which is estimated to occur in up to 40% of all patients with Type 1 or Type 2 diabetes. The concept of mitochondrial dysfunction, often involving oxidative stress, and redox disturbances, as an underlying and early response in many metabolic and degenerative diseases,42 including diabetes,43,44 suggests that targeting of processes that regulate redox status may provide a useful approach to development of novel therapies.Chronic hyperglycemia that is associated with diabetes results in increased flux of carbohydrate substrates and reducing equivalents through five metabolic pathways: (1) the polyol pathway, which leads to accumulation of sorbitol, (2) the protein kinase C (PKC) pathway, which involves accumulation of 1,2-diacylglycerol and activation of PKC, (3) the advanced glycation end-products (AGE) pathway, which involves accumulation of methylglyoxal, (4) the hexosamine pathway, which leads to accumulation of UDP-N-acetyl-D-glucosamine, and (5) mitochondrial electron transport. As illustrated in Fig. 5 (left side), the increased substrate flux and electron flow in mitochondria results in increases in release of ROS, which in turn alters redox homeostasis. The cellular response to this may depend on the extent of ROS generation: Under mild conditions, ROS may act as signaling molecules, producing compensatory upregulation of certain antioxidant genes, including those for GSH synthesis and transport;45 under conditions of higher amounts of ROS generation, mitochondrial redox changes may occur that lead to oxidative injury, including lipid peroxidation, protein alkylation or oxidation, and GSH oxidation.46
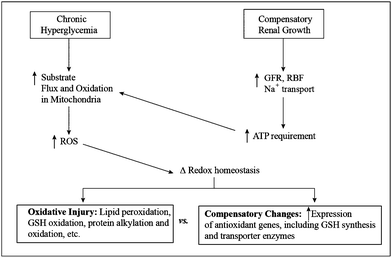 | ||
| Fig. 5 Scheme of biochemical events linking chronic hyperglycemia and compensatory renal growth to changes in mitochondrial function and redox status. Abbreviations: GFR, glomerular filtration rate; GSH, glutathione; RBF, renal blood flow; ROS, reactive oxygen species. | ||
As a starting point in assessing the role of mitochondrial redox status, and in particular, GSH status, in diabetic nephropathy, Lash and colleagues used the streptozotocin (STZ)-induced diabetic rat as a model system for Type 1 diabetes.47–49 The STZ diabetic rat typically develops frank nephropathy by 45 to 60 days after STZ injection. Accordingly, mitochondrial function, GSH redox status and transport, and susceptibility of isolated renal cortical mitochondria and primary cultures of renal proximal tubular cells to oxidants and mitochondrial toxicants were studied at two time points: 30 days after STZ injection, which is a time point at which frank nephropathy should not yet exist; 90 days after STZ injection, which is a time point after the onset of nephropathy. Rats remained hyperglycemic but were maintained on a low dose of insulin to prevent development of ketosis. The initial hypothesis was that diabetic nephropathy is associated with decreased expression and function of mitochondrial GSH transporters, leading to decreased mitochondrial GSH content, oxidative stress, and enhanced susceptibility of renal proximal tubular cells to chemically induced toxicity.47 Further, it was hypothesized that as diabetic nephropathy progressed to more advanced stages, these effects would worsen.
Study of renal mitochondrial function and GSH status in STZ diabetic rats at both 30 days and 90 days post-STZ treatment,47–49 however, revealed some surprising results and necessitated a modification in the central hypothesis. With regard to mitochondrial activity, both the rates of State 3 respiration and activities of several dehydrogenases were significantly elevated in renal mitochondria from 30-day diabetic rats as compared with those from age-matched control rats. While the mitochondrial activity results were expected, effects on GSH status were not quite as expected. Concentrations of GSH in total renal cortical homogenates, but particularly in mitochondrial homogenates, from 30-day diabetic rats were >2-fold higher than those from age-matched control rats. Rates of GSH transport into renal cortical mitochondria were higher as well. Examination of expression levels of the two carriers in kidneys from 30-day diabetic vs. age-matched control rats, however, showed decreased mRNA expression for both carriers, increased protein expression for the DIC (2.4-fold), and no difference in protein expression for the OGC. These results indicated that the changes in mitochondrial GSH transport are compensatory and due to a complex interplay of transcriptional, translational, post-translational, and kinetic changes due to the diabetic state.
Lipid peroxidation in renal mitochondria challenged with the oxidant tBH was higher in those from 30-day diabetic rats than those from age-matched control rats. Both basal and oxidant-stimulated ROS levels in proximal tubular cells, as measured with fluorescent dyes and confocal microscopy and flow cytometry, were higher in 30-day diabetic rats. These findings all suggested that even at 30 days post-STZ, when a frank state of nephropathy does not yet exist, fundamental changes have occurred in the energetics and redox status of the renal proximal tubule. Perhaps the most surprising result was that 30-day diabetic rats exhibited significant elevations as compared to age-matched control rats in urinary levels of N-acetyl-β-D-glucosaminidase (NAG), albumin, and protein, all markers of proximal tubular dysfunction. Neither blood urea nitrogen (BUN) nor serum creatinine (SCr) levels were elevated, suggesting that proximal tubular function was not markedly compromised. Additionally, histopathology analysis of renal cortical slices with hematoxylin and eosin or p-amino Shiff base revealed a modest degree of mesangial expansion and glomerular hypertrophy. Thus, the observation of significant increases in some urinary parameters and mild histological changes in kidneys of 30-day diabetic rats indicates that fundamental biochemical, physiological, and morphological changes occur in the kidneys of diabetic rats even prior to the onset of frank nephropathy.
Examination of the above mentioned biochemical parameters in 90-day STZ diabetic rats revealed that significant adaptation occurs in chronically diabetic rats. While mitochondrial State 3 respiration rates were still higher at 90 days than in age-matched control rats, the extent of elevation was less than at 30 days. Similarly, mitochondrial and total cellular GSH concentrations remained elevated at 90 days post-STZ, but the difference between levels in diabetic and control rats was not as large as at 30 days. Rates of mitochondrial GSH uptake remained elevated, mRNA expression of both the DIC and OGC were slightly elevated (1.4- and 1.1-fold, respectively), protein expression of the DIC was ∼2-fold higher in renal mitochondria from diabetic rats, and protein expression of the OGC did not differ between renal mitochondria of 90-day diabetic and age-matched control rats. From these data, Lash and colleagues49 concluded that kidneys, and specifically mitochondria, in chronically diabetic rats exhibit altered physiology and biochemistry that include both adaptive and stress response changes.
As mentioned above, the original hypothesis needed modification, based on the data from both 30-day and 90-day diabetic rats. Rather than suggesting that mitochondrial GSH was depleted due to inhibition or down-regulation of transporters, the increased concentrations and altered activities and expression of the DIC and OGC suggested that renal mitochondria from diabetic rats are in a state of oxidative stress and that although some adaptive changes occur in energetics and redox state, these changes are insufficient to maintain optimal cellular function.
Mitochondrial GSH in compensatory renal hypertrophy
Another experimental model of renal disease or chronic pathology that involves underlying changes in mitochondrial energetics and redox state and has significant clinical implications is that of compensatory renal hypertrophy (CRH) that occurs after a significant reduction in functional renal mass. Reductions in functional renal mass occur in humans during the course of aging, in severe kidney damage from diseases, or as a consequence of congenital defects. Uninephrectomy is performed in transplantation and for treatment of unilateral renal cancer that is usually secondary to a primary tumor in a different tissue. Although individuals with a single kidney typically live normally without significant or overt adverse effects, the adaptation that occurs in the remnant kidney to make up for the loss of functional tissue is associated with profound changes in renal physiology and biochemistry.50,51A commonly used experimental model to study CRH is the uninephrectomized (NPX) rat. The compensatory response, which is primarily (i.e., >85%) a cellular hypertrophy (i.e., cellular enlargement) rather than hyperplasia (i.e., cellular proliferation), is complete within 10 days of uninephrectomy and includes >50% enlargement of the overall size of the remnant kidney as compared to that prior to surgery and marked increases in glomerular filtration rate (GFR), renal blood flow (RBF), and rates of Na+ transport. This dramatic increase in workload led Schrier and colleagues52,53 to hypothesize that CRH is associated with a hypermetabolic state in renal mitochondria as a consequence of the large increase in ATP demand by renal cells. Earlier electron microscopic studies of hypertrophied kidneys demonstrated prominent mitochondrial proliferation.54 This sequence of events is illustrated in Fig. 5 (right side). We hypothesize that the biochemical, physiological, and morphological changes that occur in CRH lead to adaptive and/or oxidative changes in renal mitochondria that are similar (although not identical) to those that occur in diabetic nephropathy.
As stated above, even though individuals function reasonably well with only a single kidney, there are significant functional changes that may increase risk for development of hypertension and chronic kidney disease.55,56 There are also changes in susceptibility to nephrotoxicants, with NPX rats or renal proximal tubular cells from NPX rats exhibiting increased susceptibility to injury, as compared to normal rats or renal proximal tubular cells from normal rats, respectively. Nephrotoxicants demonstrating this altered susceptibility include analgesics,57,58 cadmium,59 and inorganic mercury.60–65 The underlying changes at the cellular level are due to changes in redox status and mitochondrial function.52,53,66–68
As with the diabetic nephropathy model discussed above, one potential hypothesis to explain the changes in renal energetics and redox status is that the mitochondrial GSH pool would be decreased, potentially due to down-regulation of transporters. This would in turn lead to enhanced oxidative stress and susceptibility to various nephrotoxicants. Somewhat surprisingly, however, a significant increase was observed in the size of the mitochondrial GSH pool, which was associated with increased rates of transport.68 Real-time polymerase chain reaction (PCR) analysis showed no difference in mRNA expression of either the DIC or OGC in renal mitochondria from control or NPX rats. Similarly, Western blot analysis showed no differences in protein expression of either the DIC or OGC. Thus, it was concluded that the increased rates of mitochondrial GSH transport in the remnant kidney of NPX rats is due to the hypermetabolic state that results in increased fluxes of intermediary metabolites, including various dicarboxylates or dicarboxylate precursors, some of which are substrates for the DIC or OGC. Thus, the increase in observed transport is due to kinetics or mass action effects. It should be noted, however, that expression or activity of several renal plasma membrane carrier proteins that function to transport organic anions, such as the organic anion transporter 1 and 3 (Oat1/3; Slc22a6/8) and multidrug resistance-associated protein 2 and 5 (Mrp2/5; Abcc2/5), is significantly higher in renal proximal tubules of NPX rats as compared to those of control rats.69,70
Modulation of mitochondrial GSH transport as a potential therapeutic approach
After identifying the carrier proteins responsible for mitochondrial GSH transport, demonstrating the variability in their expression and/or activity under different toxicological or pathological conditions, and modulating expression in an immortalized cell line, the next stage is to apply this same principle in an in vitro disease model and demonstrate that the renal toxicity or cellular dysfunction is normalized by the increased levels of mitochondrial GSH. The underlying concept, as stated earlier, is that although GSH itself or precursors of GSH can be administered to increase intracellular GSH concentrations, such an approach will only produce a transient increase in GSH concentrations and that the altered set-point will eventually re-establish levels of GSH that are insufficient for optimal function. To get around this problem, we proposed that overexpression of the DIC and/or OGC will produce a long-term increase in mitochondrial GSH concentrations by raising the set-point, thereby overcoming the limitations of the pharmacological approach.Initial studies were conducted in primary cultures of renal proximal tubular cells from 30-day STZ diabetic rats (L.H. Lash, D.A. Putt, and B. Benipal, unpublished data). As indicated above, these primary cell cultures retain the altered phenotype of proximal tubular cells in vivo and can thus be used as a convenient in vitro model to study diabetic nephropathy.48 Similarly, primary cultures of proximal tubular cells from the remnant kidney of NPX rats were shown to retain the altered phenotype of proximal tubular cells in vivo from NPX rats.65
Lipofectamine LTX (Invitrogen; Carlsbad, CA, USA) was used to transfect renal proximal tubular cells from 30-day diabetic rats with the cDNA for either the DIC or OGC. Both transfections produced substantial increases in mRNA expression of the DIC or OGC, respectively. To assess the toxicological impact of carrier overexpression, the sensitivity of transfected cells to cytotoxicity induced by the mitochondrial inhibitor antimycin A (AA; 1 and 10 μM) using lactate dehydrogenase (LDH) release was determined (Fig. 6). Proximal tubular cells were preincubated for 1 h with 1 mM GSH and were then incubated for 1 h with either cell culture medium (None) or antimycin A (AA: 1 or 10 μM). After incubations, release of LDH was measured as an index of cellular necrosis.
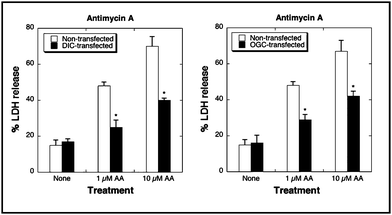 | ||
| Fig. 6 Overexpression of the DIC (left panel) or OGC (right panel) protects renal proximal tubular cells from diabetic rats from cytotoxicity induced by antimycin A. | ||
Proximal tubular cells overexpressing either carrier were significantly protected from AA-induced LDH release, supporting the viability of this experimental approach for future development of a novel therapy that can be applied, first in vivo in an animal model, and then in the clinic in patients.
Conclusions
This brief review has emphasized the fundamental importance of GSH in renal mitochondria, in both toxicological and disease states. GSH is present in all aerobic cells and is compartmentalized, with many cells having been demonstrated to possess a distinctly regulated mitochondrial pool. Because GSH synthesis appears to occur exclusively in the cytoplasm, the mitochondrial pool must derive from the function of inner membrane transporters, which exchange GSH for either inorganic phosphate or 2-OG. Although earlier studies had characterized the activity of mitochondrial GSH transport during different energetic states, identification of the function of specific carrier proteins was needed to enable a more complete understanding and manipulation of the transport process. Two organic anion carriers on the mitochondrial inner membrane of renal cortical epithelial cells, the DIC and OGC, were identified as being responsible for transporting GSH into the mitochondrial matrix. This knowledge enabled genetic manipulation of the carriers to probe their function in mitochondrial energetics and redox status. Further, this also enabled investigation of these carriers as potential therapeutic targets for treatment of exposures to oxidants or mitochondrial toxicants or various disease states that are characterized by mitochondrial dysfunction.Studies in NRK-52E cells, a stable renal cell line derived from the rat proximal tubule, demonstrated the ability of overexpression of the DIC or OGC to protect these cells from oxidant or mitochondrial toxicant-induced toxicity. Further, overexpression of a mutant OGC, which possesses <25% activity of the WT carrier, provided no protection, indicating that a threshold level of mitochondrial GSH is needed to maintain redox homeostasis and prevent oxidant-induced injury. Studies in two disease models, diabetic nephropathy and CRH, demonstrated compensatory up-regulation of mitochondrial GSH transporters that was insufficient to prevent oxidative stress and enhanced susceptibility of the renal proximal tubule to chemically induced cytotoxicity. Further, preliminary studies in an in vitro model of diabetic nephropathy, primary cultures of renal proximal tubular cells from STZ diabetic rats, suggested that overexpression of the DIC or OGC may be a promising therapeutic approach.
Examination of the various transport pathways for GSH in the renal proximal tubule (Fig. 7) highlights the interactions of GSH with dicarboxylate and intermediary metabolism. 2-OG functions as a co-substrate for the OGC on the mitochondrial inner membrane, for both Oat1 and Oat3 on the basolateral plasma membrane (BLM), for the sodium-dicarboxylate carrier 3 (NaC3; Slc13a3) on the BLM, and potentially for the organic anion transporting polypeptide 1a1 (Oatp1a1; Slco1a1) on the brush-border plasma membrane (BBM). From this observation, one can readily see how changes in cellular energetics can influence both cellular and mitochondrial GSH status and, therefore, redox status.
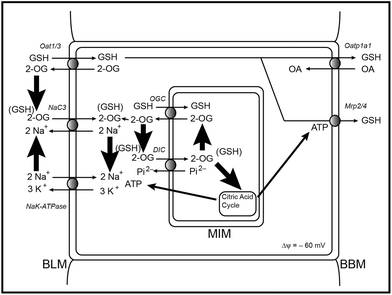 | ||
| Fig. 7 Pathways of GSH transport in the renal proximal tubule, illustrating the interactions with dicarboxylates and intermediary metabolism. Abbreviations: 2-OG, 2-oxoglutarate; BBM, brush-border plasma membrane; BLM, basolateral plasma membrane; DIC, dicarboxylate carrier; GSH, glutathione; MIM, mitochondrial inner membrane; Mrp2/4, multidrug resistance-associated protein 2/4; NaC3, sodium-dicarboxylate carrier 3; OA, organic anion; Oat1/3, organic anion transporter 1/3; Oatp1a1, organic anion transporting polypeptide 1a1; OGC, 2-oxoglutarate carrier. | ||
Acknowledgements
Research support for some of the studies summarized in this review from the author's laboratory was provided by the National Institute of Diabetes and Digestive and Kidney Diseases (R01-DK40725) and the Department of Defense Peer-Reviewed Medical Research Program (Grant PR64340; contract number W81XWH-07-1-0453).Notes and references
- J. G. de la Asuncion, A. Millan, R. Pla, L. Bruseghini, A. Esteres, F. V. Pallardo, J. Sastre and J. Vina, FASEB J., 1996, 10, 333 CAS.
- J. M. Esteve, J. Mompo, J. Garcia de la Asuncion, J. Sastre, M. Asensi, J. Boix, J. R. Vina and F. V. Pallardo, FASEB J., 1999, 13, 1055 CAS.
- B. V. Chernyak and P. Benardi, Eur. J. Biochem., 1996, 238, 623 CAS.
- P. Constantini, B. V. Chernyak, V. Petronilli and P. Bernardi, J. Biol. Chem., 1996, 271, 6746 CrossRef.
- A. P. Halestrap, K.-Y. Woodfield and C. P. Connern, J. Biol. Chem., 1997, 272, 3346 CrossRef CAS.
- A. Masini, D. Ceccarelli, T. Trenti, D. Gallesi and U. Muscatello, Biochim. Biophys. Acta, 1992, 1101, 84 CAS.
- P. Marchetti, D. Decaudin, A. Macho, N. Zamzami, T. Hirsch, S. A. Susin and G. Kroemer, Eur. J. Immunol., 1997, 27, 289 CrossRef CAS.
- C. A. Hinchman and N. Ballatori, Biochem. Pharmacol., 1990, 40, 1131 CrossRef CAS.
- L. H. Lash, Toxicol. Appl. Pharmacol., 2005, 204, 329 CrossRef CAS.
- M. J. Meredith and D. J. Reed, J. Biol. Chem., 1982, 257, 3747 CAS.
- R. G. Schnellmann and L. J. Mandel, Biochem. Biophys. Res. Commun., 1985, 133, 1001 CrossRef CAS.
- L. H. Lash, T. M. Visarius, J. M. Sall, W. Qian and J. J. Tokarz, Toxicology, 1998, 130, 1 CrossRef CAS.
- O. W. Griffith and A. Meister, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 4468 CrossRef.
- T. B. McKernan, E. B. Woods and L. H. Lash, Arch. Biochem. Biophys., 1991, 288, 653 CrossRef CAS.
- D. P. Jones, L. H. Lash, in Mitochondrial Dysfunction: Methods in Toxicology, Introduction, ed. L. H. Lash and D. P. Jones, Academic Press, San Diego, 1993, vol. 2, pp. 1–7 Search PubMed.
- L. H. Lash, Chem.-Biol. Interact., 2006, 163, 54 CrossRef CAS.
- L. Eldjarn and J. Bremer, Acta Chem. Scand., 1963, 17, S59 CrossRef CAS.
- F. E. Hunter Jr., A. Scott, P. E. Hoffsten, J. M. Gebicki, J. Weinstein and A. Schneider, J. Biol. Chem., 1964, 239, 614 CAS.
- W. D. Cash and M. Gardy, J. Biol. Chem., 1965, 240, PC3450 CAS.
- P. C. Jocelyn and A. Kamminga, Biochim. Biophys. Acta, 1974, 343, 356 CrossRef CAS.
- P. C. Jocelyn, Biochim. Biophys. Acta, 1975, 369, 427 Search PubMed.
- K. Kurosawa, N. Hayashi, N. Sato, T. Kamada and K. Tagawa, Biochem. Biophys. Res. Commun., 1990, 167, 367 CrossRef CAS.
- J. Martensson, J. C. K. Lai and A. Meister, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 7185 CrossRef CAS.
- J. C. Fernandez-Checa, C. Garcia-Ruiz, M. Ookhtens and N. Kaplowitz, J. Clin. Invest., 1991, 87, 397 CrossRef CAS.
- R. G. Schnellmann, Life Sci., 1991, 49, 393 CrossRef CAS.
- M. Klingenberg, Methods Enzymol., 1979, 56, 245 CrossRef CAS.
- F. Palmieri, F. Bisaccia, L. Capobianco, V. Dolce, G. Fiermonte, V. Iacobazzi, C. Indiveri and L. Palmieri, Biochim. Biophys. Acta, 1996, 1275, 127 Search PubMed.
- F. Palmieri, Pfluegers Arch., 2004, 447, 689 CrossRef CAS.
- Z. Chen and L. H. Lash, J. Pharmacol. Exp. Ther., 1998, 285, 608 CAS.
- Z. Chen, D. A. Putt and L. H. Lash, Arch. Biochem. Biophys., 2000, 373, 193 CrossRef CAS.
- O. Coll, C. Garcia-Ruiz, N. Kaplowitz and J. C. Fernandez-Checa, Hepatology, 2003, 38, 692 CrossRef CAS.
- Q. Zhong, D. A. Putt, F. Xu and L. H. Lash, Arch. Biochem. Biophys., 2008, 474, 119 CrossRef CAS.
- C. K. Kamga, S. X. Zhang and Y. Wang, Am. J. Physiol., 2010, 299, C497 CrossRef CAS.
- H. M. Wilkins, K. Marquardt, L. H. Lash and D. A. Linseman, Free Radical Biol. Med., 2012, 52, 410 CrossRef CAS.
- M. L. Circu, C. Rodriguez, R. Maloney, M. P. Moyer and T. Y. Aw, Free Radical Biol. Med., 2008, 44, 768 CrossRef CAS.
- L. H. Lash, D. A. Putt and L. H. Matherly, J. Pharmacol. Exp. Ther., 2002, 303, 476 CrossRef CAS.
- F. Xu, D. A. Putt, L. H. Matherly and L. H. Lash, J. Pharmacol. Exp. Ther., 2006, 316, 1175 CrossRef CAS.
- L. H. Lash, D. A. Putt, S. E. Hueni, W. Cao, F. Xu, S. J. Kulidjian and J. P. Horwitz, Biochem. Pharmacol., 2002, 64, 1533 CrossRef CAS.
- L. H. Lash and M. W. Anders, Mol. Pharmacol., 1987, 32, 549 CAS.
- F. Xu, I. Papanayotou, D. A. Putt, J. Wang and L. H. Lash, Biochem. Pharmacol., 2008, 76, 552 CrossRef CAS.
- L. H. Lash, J. C. Parker and C. S. Scott, Environ. Health Perspect., 2000, 108(Suppl 2), 225 CrossRef CAS.
- D. C. Wallace, Annu. Rev. Genet., 2005, 39, 359 CrossRef CAS.
- M. Brownlee, Nature, 2001, 414, 813 CrossRef CAS.
- A. P. Rolo and C. M. Pameira, Toxicol. Appl. Pharmacol., 2006, 212, 167 CrossRef CAS.
- D. P. Jones, Am. J. Physiol., 2008, 295, C849 CrossRef CAS.
- D. P. Jones, Chem.-Biol. Interact., 2006, 163, 38 CrossRef CAS.
- Q. Zhong and L. H. Lash, Nephroprevention, 2007, 2 Search PubMed http://www.nephroprevention.org/.
- Q. Zhong, S. R. Terlecky and L. H. Lash, Toxicol. Appl. Pharmacol., 2009, 241, 1 CrossRef CAS.
- D. A. Putt, Q. Zhong and L. H. Lash, Toxicol. Appl. Pharmacol., 2012, 258, 188 CrossRef CAS.
- L. Fine, Kidney Int., 1985, 29, 619 CrossRef.
- D. G. Shirley and S. J. Walter, Kidney Int., 1991, 40, 62 CrossRef CAS.
- D. C. H. Harris, L. Chan and R. W. Schrier, Am. J. Physiol., 1988, 254, F267 CAS.
- J. I. Shapiro, N. Elkins, O. K. Reiss, G. Suleymanlar, H. Jin, R. W. Schrier and L. Chan, Kidney Int., 1994, 45(Suppl), S100 CAS.
- H. A. Johnson and F. Amendola, Am. J. Pathol., 1969, 54, 35 CAS.
- T. H. Hostetter, Annu. Rev. Physiol., 1995, 57, 263 CrossRef CAS.
- B. L. Kasiske, J. Z. Ma, T. A. Louis and S. K. Swan, Kidney Int., 1995, 48, 814 CrossRef CAS.
- E. A. Molland, Am. J. Pathol., 1976, 120, 43 CAS.
- M. A. Henry, R. S. Sweet and J. D. Tange, Am. J. Pathol., 1983, 139, 23 CAS.
- R. K. Zalups, R. M. Gelein and M. G. Cherian, J. Pharmacol. Exp. Ther., 1992, 262, 1256 CAS.
- M. T. Houser and W. O. Berndt, Toxicol. Appl. Pharmacol., 1986, 83, 506 CrossRef CAS.
- R. K. Zalups and G. L. Diamond, Virchows Arch. B, 1987, 53, 336 CrossRef CAS.
- R. K. Zalups, Toxicol. Appl. Pharmacol., 1997, 143, 366 CrossRef CAS.
- L. H. Lash and R. K. Zalups, J. Pharmacol. Exp. Ther., 1992, 261, 819 CAS.
- L. H. Lash, D. A. Putt and R. K. Zalups, J. Pharmacol. Exp. Ther., 1999, 291, 492 CAS.
- L. H. Lash, D. A. Putt and R. K. Zalups, Toxicol. Sci., 2006, 94, 417 CrossRef CAS.
- K. A. Nath, A. J. Croattm and T. H. Hostetter, Am. J. Physiol., 1990, 258, F1354 CAS.
- L. H. Lash and R. K. Zalups, Arch. Biochem. Biophys., 1994, 309, 129 CrossRef CAS.
- L. H. Lash, D. A. Putt, S. J. Horky III and R. K. Zalups, Biochem. Pharmacol., 2001, 62, 383 CrossRef CAS.
- B. Benipal and L. H. Lash, Biochem. Pharmacol., 2011, 81, 295 CrossRef CAS.
- L. H. Lash, S. E. Hueni, D. A. Putt and R. K. Zalups, Toxicol. Sci., 2005, 88, 630 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |