Formation of a supramolecular chromophore: a spectroscopic and theoretical study†
Andreas
Bernet
ab,
Rodrigo Q.
Albuquerque
*bc,
Marina
Behr
ab,
Sebastian T.
Hoffmann
d and
Hans-Werner
Schmidt
*ab
aMacromolecular Chemistry I, University of Bayreuth, Bayreuth, 95440, Germany. E-mail: hans-werner.schmidt@uni-bayreuth.de; Fax: +49 921 553206; Tel: +49 921 55 3200
bBayreuth Institute for Macromolecular Research (BIMF), University of Bayreuth, Bayreuth, 95440, Germany
cTheoretical Physics IV, University of Bayreuth, Bayreuth, 95440, Germany. E-mail: rqa_ufpe@yahoo.com; Fax: +49 921 55 3223; Tel: +49 921 55 3363
dExperimental Physics II, University of Bayreuth, Bayreuth, 95440, Germany
First published on 22nd November 2011
Abstract
Spectroscopic and theoretical investigations of the self-assembly of a particular 1,3,5-benzene trisamide-based low molecular weight hydrogelator are described. This trisamide is pH-sensitive, and surprisingly forms a photoluminescent supramolecular hydrogel. Controlled gel formation in combination with the luminescence properties allows studying the self-assembly process in detail. The experimental results are confirmed by Density Functional Theory (DFT) calculations, revealing that the photoluminescence originates from the formation of a supramolecular chromophore.
The interest in multichromophoric assemblies has grown in the last few years, due to their potential applications as advanced functional materials.1 A fascinating bottom-up approach to realize supramolecular multichromophore systems is based on the self-assembly of low molecular weight organic compounds. Fine-tuning of the underlying molecular structure allows for a guided self-assembly into hierarchical superstructures. One particularly interesting structural motif of a supramolecular self-assembly system is based on benzene 1,3,5-tricarboxamide (BTA) derivatives.2 BTA-based molecules self-assemble into supramolecular columns in solution and in solid state3 and form liquid crystalline phases and well-defined nanostructures.4 This enables applications as thickening and gelation agents,5 nucleation and clarifying agents6 and electret additives7 for thermoplastic polymers.
Here, we present the self-assembly of the polar BTA derivative 1 (Fig. 1a) in aqueous media. We found that 1 featuring three p-carboxylphenyl side arms is one of the very rare examples8 of a BTA derivative that is able to form supramolecular hydrogels. The resulting gels are fully pH-reversible, but thermostable up to 100 °C even at the critical gelation concentration of 2 g·L−1. To the best of our knowledge, this is the only thermostable example of a supramolecular pH-reversible hydrogel system. The outstanding properties of the hydrogels will be reported in detail elsewhere.
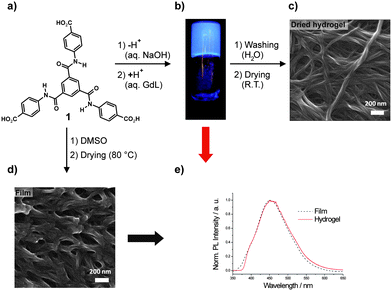 | ||
| Fig. 1 (a) Chemical structure of the investigated 1,3,5-benzene trisamide 1 (GdL: glucono-delta-lactone). (b) Photograph of a macroscopic hydrogel sample of 10 g·L−1 of 1 under UV irradiation (λexc = 366 nm). (c) SEM micrograph of the supramolecular aggregates of 1 from dried hydrogel and (d) as obtained by drop-casting from DMSO solution. (e) PL spectra of 1 in the film and in the hydrogel state (λexc = 330 nm). The corresponding absorption spectra are shown in the ESI (Fig. S2†). | ||
We also discovered that the assemblies of 1 present in the bulk and in the gel state exhibit a strong blue emission upon UV irradiation (Fig. 1b). Although 1 has been reported to be used for the production of branched polyamides9 and metal–organic frameworks,10 we were the first to discover its ability to form supramolecular hydrogels and their unexpected photoluminescence (PL) properties. In the literature, a single BTA-derived organogelator that features photoluminescence in the gel state in aprotic organic solvents is reported.11 It was concluded that the PL activity arises from molecular aggregation via H-bonding, but no further detailed explanation of the origin of the PL was given.
Herein, we investigate the self-assembly of 1 leading to a photoluminescent hydrogel. Featuring three carboxy moieties, the aggregation behavior of 1 in aqueous environment is controllable by changing the pH. When 1 is mixed with aqueous NaOH solution, it is transferred into the corresponding sodium salt 1Na which is readily water-soluble. Upon acidification the carboxylates of 1Na get protonated. This reduces the solubility, enables self-assembly and finally leads to the formation of a macroscopic hydrogel (Fig. 1b). Fig. 1c shows a SEM image of the dried hydrogel exhibiting a fibrillar three-dimensional network morphology. A different, but also nanofibrillar morphology is formed by casting a solution of 1 in DMSO and subsequent drying (Fig. 1d).
The steady-state PL spectra of 1 as dried film and in the hydrogel are shown in Fig. 1e. Although the morphologies obtained from the dried hydrogel and from the film sample look different, the obtained emission spectra are very similar. This indicates that the respective emitting excited states are the same, and that the morphological difference is only caused by different hierarchical superstructures of one common underlying assembly motif, caused by different conditions under which self-assembly of 1 takes place. Indeed, XRD spectra of both specimens (see ESI, Fig. S4†) indicate that in the film sample there is a much higher long-range order than in the xerogel sample. It has to be noted that PL was only observed in the presence of the supramolecular structures. In DMSO solutions at concentrations up to 10 g·L−1 of 1, no PL was detectable with a standard PL setup.
In order to investigate the formation of the luminescent supramolecular hydrogel, it is necessary to decrease the pH slowly in a continuous and controlled manner. This can be accomplished by mixing an aqueous solution of 1Na with glucono-delta-lactone (GdL).12 The hydrogel of 1 is then gradually formed, which enables recording the luminescence spectra over time (Fig. 2).13 Before the addition of GdL, no PL is observed (a). 2.5 min after addition, a first peak at ca. 380 nm is visible (b). This peak can be retrieved throughout the following absorption spectra (c to g). At 7.5 min, a second peak evolves at ca. 450 nm and is increasing over time showing a slight red-shift (c to g).
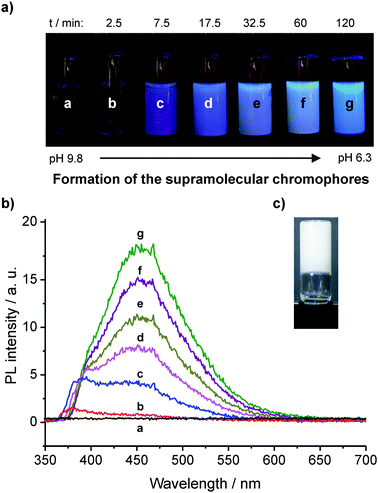 | ||
| Fig. 2 (a) Photographs of a macroscopic sample of an aqueous solution of 10 g·L−1 of 1Na before (a) and after (b to g) addition of GdL (images were recorded under UV irradiation, λexc = 366 nm. Differences in color are caused by scattering effects and automatic white balance of the camera). (b) Corresponding PL spectra (λexc = 300 nm. The noise is caused by the fast scanning speed that was chosen in order to avoid significant changes of the spectra during acquisition). (c) Photograph of inverted hydrogel sample after 24 h (although the pH value continuously decreases reaching a value of ca. 4.2 after 4 days, there are no significant changes in the PL spectra compared to the sample after 2 h (g)). | ||
Before the addition of GdL, the highly charged 1Na is molecularly dissolved due to electrostatic repulsion as indicated by the absence of significant scattering signal in DLS experiments. After the addition of GdL, protons are released by the hydrolysis of GdL. From a statistical point of view, it is reasonable to assume that at first only one carboxylate per molecule of 1Na gets protonated. As this lowers the solubility, the partially protonated 1Na molecules start aggregating upon encountering each other. Two aggregation modes are possible: dimerization of two molecules via the carboxylic groups leads to “side-by-side” dimers (ESI, Fig. S7†), whereas the stacking of two molecules driven by triple hydrogen bonding of the amide groups and π–π interaction of the aromatic cores yields energetically more favorable “on-top” dimers (ESI, Fig. S8†). Over time, with the further decrease of pH more and more of the initially formed dimers can aggregate into more extended columnar superstructures.
So far, only a few computational studies on BTA derivatives have been reported,2,14 and the majority of the mentioned studies is focusing on the behavior of BTAs in organic solvents and polymer melts. Aiming to understand how the self-assembly of 1 influences the optical properties of the resulting aggregates, DFT and time-dependent DFT (TDDFT) calculations were carried out for the monomer, dimer, trimer and tetramer forms of 1.15 The optimized geometries of monomer and tetramer (for dimers and trimer see ESI, Fig. S7–S9†) together with the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) are shown in Fig. 3a.
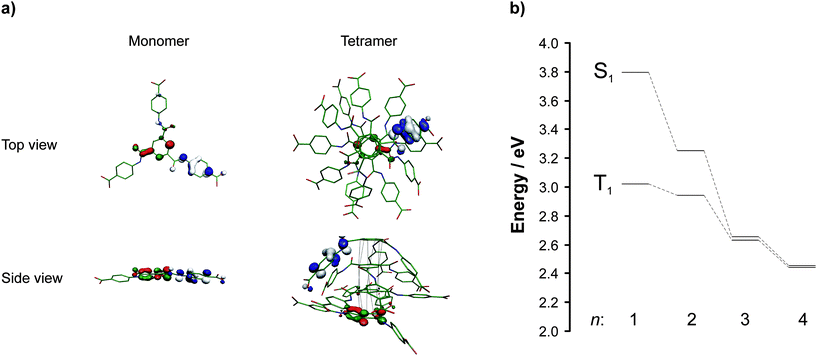 | ||
| Fig. 3 (a) DFT optimized geometries of 1 as a monomer and a tetramer (top view and side view are shown) with the respective frontier molecular orbitals HOMO (blue/white) and LUMO (red/green). The atom labels are: carbon (green), nitrogen (blue), and oxygen (red). The hydrogens are omitted for clarity. The black dotted lines represent the stacking of the aromatic cores. (b) TDDFT calculated energies of the excited singlet (S1) and triplet (T1) states of aggregates of 1.The n values refer to the number of monomer units of 1. | ||
In the case of dimer, trimer, and tetramer, columnar stacking of monomer units takes place due to π–π interaction and triple hydrogen bonding of the benzene trisamide core. The presence of hydrogen-bonded amide units in dried hydrogel and bulk samples of 1 was experimentally proven by FT-IR (ESI, Fig. S5†). The calculated core distances are 3.37 Å both for the dimer and trimer and 3.28 Å for the tetramer, respectively. Although the dispersion correction seems to overestimate the π–π interaction, the calculated values are still in reasonable agreement with the results of the X-ray analysis of a dried hydrogel sample (3.47 Å, see ESI, Fig. S4†) and in a typical range for π–π interacting systems. In addition to the interactions of the aromatic cores, π–π interactions of the phenylene units of the side arms can be recognized, leading to a bending of the aromatic side arms. However, it is reasonable to assume that this edge effect will be evened out when increasing the number of monomer units in a supramolecular column. Additionally, hydrogen bonding between carboxylic groups of different columns is likely to occur. This also will counteract the bending of the side arms. Collectively, the computed noncovalent intermolecular interactions lead to a columnar stacking of monomers of 1 with a twist angle of ca. 60°. This fits very well in the packing model of BTA derivatives proposed by Meijer and co-workers.2
The calculated S1 and T1 excited states of the monomer and the “on-top” dimer, trimer and tetramer are basically localized on the central aromatic cores, which are less exposed to solvent molecules, and shielding effect becomes more pronounced upon increasing the number n of monomer units of 1. This may explain the very similar emission spectra observed for supramolecular aggregates of 1 in different chemical environments as in the hydrogel and in the thin film. However, it has to be noted that the aggregates in the film and in the hydrogel are hierarchical superstructures of 1 with much larger n values than the calculated aggregates. In Fig. 3b, the calculated energy levels for aggregates of 1 having different n values are shown.
From monomer to tetramer, the energy levels are lowered, and the singlet–triplet splitting decreases. The strong hydrogen bonds between complementary amide groups inside the same supramolecular column, as well as π–π interactions, cause a considerable red shift in the energies of those excited states. This reflects the experimental findings during the controlled gelation/PL experiments. We assign the first visible peak in the PL spectrum to the formation of an “on-top” dimer. The second detected peak and its red-shift over time fit quite well to the build-up of aggregates of 1 with an increasing n.
A “side-by-side” dimer of 1 as the origin of the second peak is unlikely, as TDDFT calculations show that the corresponding energy values of S1 and T1 are comparable to those found for the monomer (see ESI, Table S1†), meaning that the experimentally observed red shift can only be reproduced by the calculations assuming the formation of the “on-top” dimer. Furthermore, we can also conclude that a bundling of several columns caused by dimerization of peripheral carboxylic groups of different columns has a negligible influence on the energy values of the underlying structures. This type of peripheral interactions leads to a rigidification of the whole supramolecular structure, thus resulting in smaller non-radiative rates and causing the well-known aggregation-induced emission enhancement.16
Additionally, DFT reveals that S1 and T1 computed for all aggregation forms of 1 are mostly described by HOMO → LUMO excitations. These states have a strong charge transfer (CT) character, the charge being transferred from the peripheral side groups to the central aromatic cores.17 Although HOMO and LUMO show only poor overlap, transitions to higher-lying excited states, which are described by (HOMO − x) → (LUMO + y) excitations, do possess much larger oscillator strength and still exhibit CT character. After absorption, energy transfer to defects may also play a role in the emission.18
In agreement with the literature, all amide groups within a supramolecular column are pointing to the same direction, giving rise to a strong macrodipole.19 This may explain the CT character of the excited states of supramolecular aggregates of 1, as a spatial charge separation can stabilize the macrodipole. However, it is important to remember that the structure shown in Fig. 3 is a very simplified model that helps to understand the experimental results. Therefore molecular dynamics simulations are necessary to achieve a more complete representation of the aggregates, since a great number of interactions are expected to be present in the film and in the gel.
Conclusions
The solid-state luminescence of 1 in combination with the pH-controllable self-aggregation in aqueous media has proved to be highly useful to investigate the self-assembly process of the widely used BTA motif. DFT and experimental results strongly suggest that the blue luminescence recorded for the film and the hydrogel of 1 arises exclusively from supramolecular columnar stacks with a large number of monomers. The strong CT character of the calculated excited states, together with their well-shielded electron densities, suggest that 1 is an excellent candidate to be used as a model system for new functional supramolecular materials like non-covalent molecular wires.Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft (DFG) in the frame of Research Training Group GRK 1640 and Priority Program SPP 1259 is gratefully acknowledged. Synthesis of compound 1 was carried out within the Collaborative Research Centre SFB 840 (project B4). The German-Israeli Foundation is acknowledged for financial support. We thank B. Brunner for performing the elemental analysis (Prof. A. Jess, Department of Chemical Engineering, University of Bayreuth) and Martina Heider for the SEM experiments (Bayreuth Institute for Macromolecular Research, University of Bayreuth). We gratefully acknowledge Prof. Anna Köhler (Experimental Physics II, University of Bayreuth) for fruitful discussions.Notes and references
- E. Collini and G. D. Scholes, Science, 2009, 323, 369 CrossRef CAS; B. Bodenant, F. Fages and M. H. Delville, J. Am. Chem. Soc., 1998, 120, 7511 CrossRef CAS; O. Mirzov, R. Bloem, P. R. Hania, D. Thomsson, H. Z. Lin and I. G. Scheblykin, Small, 2009, 5, 1877 CrossRef CAS; W. T. Wiesler, J. T. Vazquez and K. Nakanishi, J. Am. Chem. Soc., 1987, 109, 5586 CrossRef CAS; J. Hofkens, M. Maus, T. Gensch, T. Vosch, M. Cotlet, F. Kohn, A. Herrmann, K. Mullen and F. De Schryver, J. Am. Chem. Soc., 2000, 122, 9278 CrossRef CAS; J. Hofkens, M. Cotlet, T. Vosch, P. Tinnefeld, K. D. Weston, C. Ego, A. Grimsdale, K. Mullen, D. Beljonne, J. L. Bredas, S. Jordens, G. Schweitzer, M. Sauer and F. De Schryver, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13146 CrossRef CAS; J. W. Verhoeven, J. Photochem. Photobiol., C, 2006, 7, 40 CrossRef CAS.
- M. P. Lightfoot, F. S. Mair, R. G. Pritchard and J. E. Warren, Chem. Commun., 1999, 19, 1945 RSC; P. J. M. Stals, M. M. J. Smulders, R. Martín-Rapún, A. R. A. Palmans and E. W. Meijer, Chem.–Eur. J., 2009, 15, 2071 CrossRef CAS.
- P. J. M. Stals, J. C. Everts, R. de Bruijn, I. A. W. Filot, M. M. J. Smulders, R. Martín-Rapún, E. A. Pidko, T. F. A. de Greef, A. R. A. Palmans and E. W. Meijer, Chem.–Eur. J., 2010, 16, 810 CAS; T.-Q. Nguyen, R. Martel, P. Avouris, M. L. Bushey, L. Brus and C. Nuckolls, J. Am. Chem. Soc., 2004, 126, 5234 CrossRef CAS.
- C. Xue, F. Ilhan, S. Jin, S. Z. D. Cheng, M. A. Meador and R. K. Eby, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 2004, 45, 820 Search PubMed; P. J. M. Stals, J. F. Haveman, R. Martín-Rapún, C. F. C. Fitié, A. R. A. Palmans and E. W. Meijer, J. Mater. Chem., 2009, 19, 124 RSC; Y. Matsunaga, N. Miyajima, Y. Nakayasu, S. Sakai and M. Yonenaga, Bull. Chem. Soc. Jpn., 1988, 61, 207.
- Y. Yasuda, E. Iishi, H. Inada and Y. Shirota, Chem. Lett., 1996, 25, 575 CrossRef; K. Hanabusa, C. Koto, M. Kimura, H. Shirai and A. Kakehi, Chem. Lett., 1997, 26, 429 CrossRef; S. J. Lee, C. R. Park and J. Y. Chang, Langmuir, 2004, 20, 9513 CrossRef CAS; M. de Loos, J. H. van Esch, R. M. Kellogg and B. L. Feringa, Tetrahedron, 2007, 63, 7285 CrossRef CAS; J. J. van Gorp, J. A. J. M. Vekemans and E. W. Meijer, J. Am. Chem. Soc., 2002, 124, 14759 CrossRef CAS.
- M. Blomenhofer, S. Ganzleben, D. Hanft and H.-W. Schmidt, Macromolecules, 2005, 38, 3688 CrossRef CAS; M. Kristiansen, P. Smith, H. Chanzy, C. Baerlocher, V. Gramlich, L. McCusker, T. Weber, P. Pattison, M. Blomenhofer and H.-W. Schmidt, Cryst. Growth Des., 2009, 9, 2556 CrossRef CAS; F. Abraham, S. Ganzleben, D. Hanft, P. Smith and H.-W. Schmidt, Macromol. Chem. Phys., 2010, 211, 171 Search PubMed; F. Abraham and H.-W. Schmidt, Polymer, 2010, 51, 913 Search PubMed; M. Kristiansen, A. Greß, P. Smith, D. Hanft and H.-W. Schmidt, Polymer, 2006, 47, 249 Search PubMed.
- N. Mohmeyer, N. Behrendt, X. Zhang, P. Smith, V. Altstädt, G. M. Sessler and H.-W. Schmidt, Polymer, 2007, 48, 1612 Search PubMed.
- D. K. Kumar, D. A. Jose, P. Dastidar and A. Das, Chem. Mater., 2004, 16, 2332 CrossRef CAS; N. Shi, H. Dong, G. Yin, Z. Xu and S. Li, Adv. Funct. Mater., 2007, 17, 1837 CrossRef CAS; N. Shi, G. Yin, M. Han and Z. Xu, Colloids Surf., B, 2008, 66, 84 CrossRef CAS.
- For example see: S. M. Aharoni, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 1989, 30, 125 Search PubMed; S. M. Aharoni and S. F. Edwards, Macromolecules, 1989, 22, 3361 Search PubMed; M. E. Cosulich, S. Russo, S. Pasquale and A. Mariani, Polymer, 2000, 41, 4951 CrossRef CAS.
- X. Song, Y. Zou, X. Liu, M. Oha and M. S. Lah, New J. Chem., 2010, 34, 2396 RSC; R. Ma, C. Chen, B. Sun, X. Zhao and N. Zhang, Inorg. Chem. Commun., 2011, 14, 1532 Search PubMed.
- S. Y. Ryu, S. Kim, J. Seo, Y.-W. Kim, O.-H. Kwon, D.-J. Jang and S. Y. Park, Chem. Commun., 2004, 1, 70 Search PubMed.
- D. J. Adams, M. F. Butler, W. J. Frith, M. Kirkland, M. Mullen and P. Sanderson, Soft Matter, 2009, 5, 1856 RSC; L. Chen, K. Morris, A. Laybourn, D. Elias, M. R. Hicks, A. Rodger, L. Serpell and D. J. Adams, Langmuir, 2010, 26, 5232 CrossRef CAS.
- Although the use of mineral acids (i.e. concentrated aqueous HCl) instead of GdL results in very fast hydrogel formation and a much lower final pH value, the PL spectra of the resulting hydrogel samples are comparable to those obtained with the GdL method.
- F. D. Lewis, T. M. Long, C. L. Stern and W. Liu, J. Phys. Chem. A, 2003, 107, 3254 CrossRef CAS; A. Rochefort, E. Bayard and S. Hadj-Messaoud, Adv. Mater., 2007, 19, 1992 CrossRef CAS.
- The B3LYP functional was used in combination with the dispersion correction of Grimme, in order to take into account π–π interactions: S. Grimme, J. Comput. Chem., 2004, 25, 1463 Search PubMed.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740 RSC; R. Deans, J. Kim, M. R. Machacek and T. M. Swager, J. Am. Chem. Soc., 2000, 122, 8565 CrossRef CAS; M. K. Nayak, B.-H. Kim, J. E. Kwon, S. Park, J. Seo, J. W. Chung and S. Y. Park, Chem.–Eur. J., 2010, 16, 7437 CrossRef CAS; Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC.
- Comparable conclusions were drawn regarding computational studies on other hydrogen-bonded supramolecular aggregates: A. Demenev, S. H. Eichhorn, T. Taerum, D. F. Perepichka, S. Patwardhan, F. C. Grozema, L. D. A. Siebbeles and R. Klenkler, Chem. Mater., 2010, 22, 1420 Search PubMed.
- The observed large Stokes shift may also be caused by some kind of orientational disorder inside the column (the emission then coming from defects after energy transfer along a supramolecular fiber), as shown in a theoretical work of Markovitsi and co-workers. The energy transfer mechanism of this class of compound is currently under investigation and will be subject of a full paper. See also: D. Markovitsi, Mol. Cryst. Liq. Cryst., 2003, 397, 89 Search PubMed; D. Markovitsi, S. Marguet, J. Bondkowski and S. Kumar, J. Phys. Chem. B, 2001, 105, 1299 Search PubMed; D. Markovitsi, A. Germain, P. Millié, P. Lécuyer, L. K. Gallos, P. Argyrakis, H. Bengs and H. Ringsdorf, J. Phys. Chem., 1995, 99, 1005 CrossRef CAS.
- C. F. C. Fitié, W. S. C. Roelofs, M. Kemerink and R. P. Sijbesma, J. Am. Chem. Soc., 2010, 132, 6892 CrossRef CAS; A. Sakamoto, D. Ogata, T. Shikata, O. Urakawa and K. Hanabusa, Polymer, 2006, 47, 956 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details of all optimized structures. See DOI: 10.1039/c1sm06789c |
| This journal is © The Royal Society of Chemistry 2012 |