Switchable bistable ordering and real-time alignment dynamics in wormlike micelles†
Kyle G.
Wilmsmeyer
,
Xiaolin
Zhang
and
Louis A.
Madsen
*
Department of Chemistry and Macromolecules and Interfaces Institute, Virginia Polytechnic Institute and State University, Blacksburg, VA 24061, USA. E-mail: lmadsen@vt.edu
First published on 10th November 2011
Abstract
Using a combination of rheology and magnetic resonance known as “rheo-NMR” we observe, in real time, the shear- and field-dependent dynamics of orientational order in a wormlike micelle (WLM). We construct a dynamical phase diagram, yielding a range where bistable shear-activated phase switching occurs in which WLMs are stably isotropic (> 12 h) before shearing, and yet realign with B0 after shearing. Furthermore, we extract anisotropic viscoelastic coefficients for the first time in a WLM by precisely fitting nonlinear realignment data. This deepened understanding of complex surfactant fluid dynamics lights a path toward increased materials control in advanced lubricants and coatings, drug delivery, and tissue engineering.
Cylindrical, or wormlike, micelles constitute an intermediate phase of soft matter, encompassing properties of liquid crystals, entangled polymers, and molecular amphiphiles, with applications as diverse as personal care products, oil exploration, and lubrication. Amphiphiles can associate into various supramolecular aggregates, including spheres, vesicles, lamellae, and cylinders, based on molecular functionality and the relative sizes of their hydrophobic and hydrophilic functionalities.1,2 WLMs posess unique viscoelastic, patterning, and encapsulation properties and thus represent pivotal components used in fracturing fluids in oil fields,3 personal care product viscosity modifiers,3 electrospun tissue scaffolds,4 and drug delivery agents.5 WLMs can be made polymerizable,6 and can form effective polymerization templates in which the resulting polymer retains the high aspect ratio of the micellar template and exhibits narrow polydispersity.7 WLMs have also been shown to form viscoelastic gels8 which can be adopted as scaffolds for nanomaterial manufacture. After intense experimental and theoretical scrutiny, many fundamental behaviors of WLMs evade understanding, such as: How does ordering couple to viscoelastic behavior? What is the nature of WLM entanglement?
WLM systems exhibit properties similar to polymer solutions, such as nonlinear stress-strain behavior, as the wormlike “chains” entangle with one another in analogy to conventional polymer chains, and these cylinders can additionally align to form a nematic liquid crystalline phase.1,9,10 Here, we study cetyltrimethylammonium bromide (CTAB) in the range 17–23 wt% in 2H2O, where CTAB self-assembles into cylinders of ∼ 4 nm diameter.11
We employ rheo-NMR12,13 to provide detailed molecular-scale understanding of these materials under macroscopic shear forces. Rheo-NMR is non-destructive, in situ, allows for straightforward theoretical interpretation, and does not require optically transparent samples. Shear-induced behavior such as shear banding and the shear-dependence of the isotropic-to-nematic transition have been addressed using rheo-NMR,14,15small-angle neutron scattering under shear (SANSUS),16 and theory.17 Here, we use 2H NMR spectroscopy to observe background solvent 2H2O molecules, which report on local micellar order. Previous studies involving CTAB WLMs have used 2H NMR to show spontaneous (static) magnetic alignment in the nematic phase.18 The present study allows observations of a bistable shear-induced transition not previously observed in any WLM system. Futhermore, by time-resolving the complex director realignment dynamics (from shear to magnetic alignment), and employing a continuum Leslie-Ericksen theory formalism,19,20 we extract the first measurements of Leslie-Ericksen anisotropic viscoelastic parameters for a WLM.
We monitor WLM phase alignment sensitively as a function of shear in situ, and construct a dynamical phase diagram of shear and magnetic alignment as a function of composition and temperature T. We find a broad range of composition and T over which the nematic WLMs spontaneously align with the spectrometer magnetic field (B0), or they can shear align perpendicular to B0 in our axial Couette cell. At higher T, CTAB solutions exhibit a region of isotropic behavior where they will not align with B0 or applied shear. Furthermore, we observe a novel intermediate phase that maintains long-term isotropy in a magnetic field until aligned in a perpendicular shear field. When shear is ceased, the phase realigns and stabilizes along the much weaker magnetic field. This intermediate phase region persists in either isotropic (pre-shear) or magnetically-aligned nematic (post-shear) phase for > 12 h, with transition back to the isotropic pre-shear phase accomplished by a simple temperature cycle.
Because deuterium is a spin I = 1 nucleus with a large electric quadrupole moment, its unique properties can be exploited in NMR experiments. The electric quadrupole moment of the nucleus interacts with the electric field gradient of the molecular bond in which it resides to produce a splitting in the 2H spectrum.21 In our system, the deuterons on the 2H2O probe molecules show a quadrupole splitting ΔνQ in the spectrum described by21,22
| ΔνQ = QpSP2(cosϕ) = QpρSmatrixP2(cosϕ) | (1) |
Isotropic tumbling, as in a simple liquid, causes molecules to experience all angles χ rapidly compared with the NMR (Qp) timescale, averaging the quadrupolar Hamiltonian (and thus the splitting) to zero.14 However, if a deuterated probe molecule (here, 2H2O) is present in an aligned WLM system, the motional averaging of the deuterated probe tumbling within the constraints of the micelles will be incomplete, causing the molecule to “inherit” the order of its surroundings, and exhibit a doublet in the spectrum.21–23 Thus, at a given temperature and composition with monodomain alignment, ΔνQ reports on the angle and degree of WLM matrix alignment under shear or magnetic fields. We observe only a single doublet in the spectrum during shear and B0 alignment, indicating uniaxial monodomain alignment.
NMR spectra at equilibrium show that WLMs spontaneously align with B0 over a wide range of temperatures. This alignment occurs due to the anisotropy of magnetic susceptibility (χa) of the cylindrical worms, and is attributed simply to the susceptibility difference between the WLMs and solvent coupled with the shape anisotropy of the cylinders. Field-induced orientational order is quite common in liquid crystalline materials21 and has been reported in nematic wormlike micelle systems.9,18 Alignment with B0 occurs with two timescales governed by distinct processes, with equilibrium typically reached in a few hours. The alignment dynamics follow Leslie-Ericksen continuum dynamic theory,19,20 which is the basis for our mathematical treatment below. Further, we see an increase in quadrupolar splitting values with increasing concentration, which is likely due to the 2H2O probe molecules being increasingly constrained as the WLM volume fraction rises (ρ rises).
In this WLM system, we investigate orientational order dependencies on concentration, shear rate (ranging from γ˙ = 0.01 to 130 s−1), time after initiating shear, and T. Complete alignment under shear rate γ˙ = 1 s−1 occurs significantly faster (∼ 10 s) than the spontaneous field alignment at the same T, signifying that even moderate shear forces are orders of magnitude greater than the alignment force of the B0 field. We can define a “critical shear rate” (γ˙c) at which shear and field alignment forces exactly cancel one another and provide a singlet in the spectrum. For example, with 20 wt% CTAB in 2H2O at 28 °C, we find γ˙c ∼ 0.06 s−1.
Using data from both spontaneous field alignment and shear alignment experiments, we have constructed the dynamical phase diagram shown in Fig. 1. We observe two distinct regions, which we have labeled as “field alignement” (FA) and “shear alignment” (SA). In the FA region, the WLMs align parallel to B0. In the SA region, the WLMs can be aligned perpendicular to B0, notably over a range of T higher than where they spontaneously field align. The FA direction is signified by the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of quadrupole splitting in the FAvs.SA states (−0.5ΔνQFA = ΔνQSA), following the P2(cosϕ) dependence of eqn (1) (see Fig. 2). Note that eqn (1) has a zero at ϕ = 54.7° (the so-called “magic” angle), which causes the spectrum to again manifest as a singlet. This singlet appears solely because the micellar director rotates through 54.7°, not due to the system transitioning to the isotropic phase. Under static conditions, these two cases are indistinguishable, but we can separate them easily with our dynamical rheo-NMR measurements. Within the FA region, shear alignment can still be achieved; however, in the SA-only region, field alignment will not spontaneously occur without prior stimulus. Previous phase diagrams determined by NMR and birefringence14,16,18 indicate regions of nematic, isotropic, and hexagonal behavior of the CTAB WLMs, but do not describe dynamic phase behavior induced by shear forces. The upper limit of the SA region is derived at shear alignment plateau values with γ˙ ≥ 10 s−1.
:1 ratio of quadrupole splitting in the FAvs.SA states (−0.5ΔνQFA = ΔνQSA), following the P2(cosϕ) dependence of eqn (1) (see Fig. 2). Note that eqn (1) has a zero at ϕ = 54.7° (the so-called “magic” angle), which causes the spectrum to again manifest as a singlet. This singlet appears solely because the micellar director rotates through 54.7°, not due to the system transitioning to the isotropic phase. Under static conditions, these two cases are indistinguishable, but we can separate them easily with our dynamical rheo-NMR measurements. Within the FA region, shear alignment can still be achieved; however, in the SA-only region, field alignment will not spontaneously occur without prior stimulus. Previous phase diagrams determined by NMR and birefringence14,16,18 indicate regions of nematic, isotropic, and hexagonal behavior of the CTAB WLMs, but do not describe dynamic phase behavior induced by shear forces. The upper limit of the SA region is derived at shear alignment plateau values with γ˙ ≥ 10 s−1.
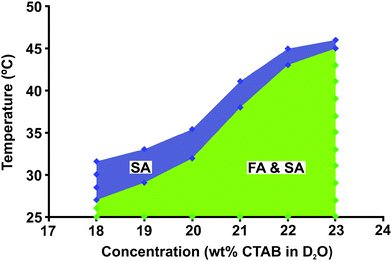 | ||
| Fig. 1 Dynamical phase diagram for CTAB in 2H2O as a function of temperature and concentration. Two regions manifest in our rheo-NMR experiments: one where field alignment occurs spontaneously along with readily attainable shear alignment (labeled FA & SA), and a higher T region where micelles do not spontaneously field align, but do shear align (labeled SA). | ||
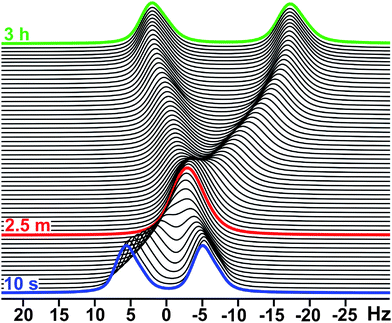 | ||
| Fig. 2 2H NMR spectra showing WLM realignment after shear cessation. Data shown for 20 wt % CTAB at 31 °C, covering 3 h after cessation of shear (γ˙ = 31.42 s−1). In accordance with eqn (1), the initial value of ΔνQ (first spectrum at t = 10 s) is approximately ½ that of the final value (after 3 h – field aligned), and there is a crossover (single peak) at t = 2.5 min when the director is along the magic angle ϕ = 54.7°. | ||
In addition to the field and shear alignment properties discussed, we find that upon raising T a few degrees above the initial FA range (e.g., to 33 °C for 20 wt% CTAB), the WLMs will align as expected under shear, but also exhibit intriguing field alignment properties after shear (see Fig. 1). Specifically, upon cessation of shear, Fig. 2 shows WLM realignment with B0, which occurs over the entire FA & SA phase region of Fig. 1, but surprisingly even at temperatures greater than the initial pre-shear field alignment range (in the SA-only region). The spectra in Fig. 2 also indicate that the transition between SA and FA occurs such that there is no breakup of domains that rotate at different rates. We do not observe broad signals or powder patterns (superpositions) in the NMR spectra on the averaging timescale of this NMR experiment (∼ 10 ms), and thus all domains rotate at the same rate during realingnment with minimal variation in angle between domains.
Fig. 3 shows a proposed WLM realignment scheme, wherein the highly entangled WLMs are broken apart by the shear forces into aligned domains along the shear direction. After shear cessation, small domains (likely encompassing shorter cylinders) can then rapidly realign with the magnetic field, and finally the small domains grow slowly into a monodomain with greater overall ordering, and likely encompassing longer worms. We note that the isotropic and field-aligned nematic phases have long-term stability (> 12 h) in the spectrometer magnetic field, and hence are bistable and switchable.
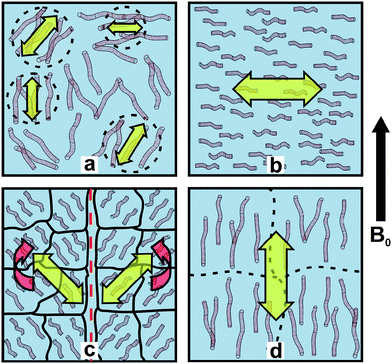 | ||
| Fig. 3 Illustration of WLM realignment motif. a, isotropic solution of WLMs prior to shear, b, WLMs under strong shear flow (ϕ = 90°), c, backflow and realignment with spectrometer field (B0) following cessation of shear, d, stable alignment with B0 (ϕ = 0°). The arrow on the right indicates the direction of the magnetic field (perpendicular to shear direction), block arrows indicate WLM director alignment, and curved arrows indicate rotation direction in the B0 field. As an example, the director is at ϕ ≈ 54.7° in part c, where the NMR splitting vanishes. | ||
We fit our spectroscopic data based on the treatments of nematic liquid crystalline polymer (LCP) systems outlined by Martins et al.19 and Kothe et al.20 The director dynamics follow that of backflow and realignment observed in similarly realigning LCPs. Because the value of χa has not yet been measured for the CTAB/2H2O or any similar WLM system, our fit utilizes a fixed estimate of 1.0 × 10−7 cgs based on measurements of other lyotropic nematics.24,25 Fit results using this estimate are excellent (see Fig. 4), and are quite sensitive to order of magnitude changes in χa.
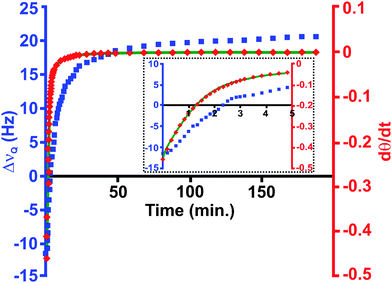 | ||
| Fig. 4 WLM realignment dynamics. Micellar director realigns with B0 after (perpendicular) shear alignment (γ˙ = 31.42 s−1). ΔνQ splittings are shown as squares and calculated dθ/dt values as diamonds. The solid line is the fit to dθ/dt, yielding viscoelastic parameters. The inset details the first 5 min of realignment. Composition is 20 wt % and T = 31 °C. | ||
Our fitting program provides values for k33/k11,26 γ1, α1, α2, and ηc. Values for ηb, α3, γ2 are derived from the Parodi relations27 and equations relating the Miesowicz viscosities28 with the Leslie coefficients.29,30Table 1 shows typical fit results for the CTAB/2H2O system. It is important to note that we observe the expected sign in all cases, however our measured parameters are 1–2 orders of magnitude larger than those reported on LCPs by Martins.19 Because these are the first reported measurements of the viscoelastic parameters for a WLM system, here we make comparisons with other systems that behave as nematic liquid crystals. Sample preparation and rheo-NMR experimental details31 can be found in electronic supplementary information (ESI†).
We have investigated phase behavior and dynamics of shear and magnetic field alignment in CTAB/2H2O WLMs as a function of composition, aligning field strength, and temperature. We observe a novel bistable shear-induced phase in which WLMs will not align along B0 until after they have been shear aligned. This work, to our knowledge, is the first report of measurements of the anisotropic viscoelastic parameters for a WLM system. We are continuing to study this system by correlating solution rheology measurements to our rheo-NMR experiments. Additionally, we are using pulsed-field-gradient NMR techniques to investigate self-diffusion behavior of the WLMs in both the aligned and unaligned state, and have begun to experiment with inclusion of molecules which reside either in the core of the micelles or in between the micelles, for potential applications as variable NMR alignment media in biological structure determination. Deep understanding of these systems will promote further design of micellar materials, and allow tailoring of viscoelastic and long-range alignment behavior for applications in tissue engineering, oil extraction, and advanced coatings and lubricants.
Acknowledgements
This material is based upon work supported in part by the U.S. Army Research Office under Grant W911NF-07-1-0452 Ionic Liquids in Electro-Active Devices (ILEAD) MURI. Acknowledgment is also made to Virginia Tech for startup funds.References
- M. E. Cates and S. J. Candau, J. Phys.: Condens. Matter, 1990, 2, 6869–6892 CrossRef CAS.
- D. F. Evans and B. W. Ninham, J. Phys. Chem., 1986, 90, 226–234 CrossRef CAS.
- J. Yang, Curr. Opin. Colloid Interface Sci., 2002, 7, 276–281 CrossRef CAS.
- M. G. McKee, J. M. Layman, M. P. Cashion and T. E. Long, Science, 2006, 311, 353–355 CrossRef CAS.
- Y. Kim, P. Dalhaimer, D. A. Christian and D. E. Discher, Nanotechnology, 2005, 16, S484–S491 CrossRef.
- S. Y. Liu, Y. I. Gonzalez, D. Danino and E. W. Kaler, Macromolecules, 2005, 38, 2482–2491 CrossRef CAS.
- L. M. Walker and D. M. Kuntz, Curr. Opin. Colloid Interface Sci., 2007, 12, 101–105 CrossRef CAS.
- M. Vasudevan, E. Buse, D. L. Lu, H. Krishna, R. Kalyanaraman, A. Q. Shen, B. Khomami and R. Sureshkumar, Nat. Mater., 2010, 9, 436–441 CrossRef CAS.
- T. M. Alam and S. K. McIntyre, Langmuir, 2008, 24, 13890–13896 CrossRef CAS.
- M. R. Lopez-Gonzalez, W. M. Holmes and P. T. Callaghan, Soft Matter, 2006, 2, 855–869 RSC.
- L. Coppola, R. Gianferri, I. Nicotera, C. Oliviero and G. A. Ranieri, Phys. Chem. Chem. Phys., 2004, 6, 2364–2372 RSC.
- A. I. Nakatani, M. D. Poliks and E. T. Samulski, Macromolecules, 1990, 23, 2686–2692 CrossRef CAS.
- P. T. Callaghan, Rep. Prog. Phys., 1999, 62, 599–670 CrossRef CAS.
- E. Fischer and P. T. Callaghan, Phys. Rev. E, 2001, 64, 6401 Search PubMed.
- R. Angelico, D. Burgemeister, A. Ceglie, U. Olsson, G. Palazzo and C. Schmidt, J. Phys. Chem. B, 2003, 107, 10325–10328 CrossRef CAS.
- E. Cappelaere, J. F. Berret, J. P. Decruppe, R. Cressely and P. Lindner, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1997, 56, 1869–1878 CrossRef CAS.
- C. Y. D. Lu, P. D. Olmsted and R. C. Ball, Phys. Rev. Lett., 2000, 84, 642–645 CrossRef CAS.
- J. S. Clawson, G. P. Holland and T. M. Alam, Phys. Chem. Chem. Phys., 2006, 8, 2635–2641 RSC.
- A. F. Martins, P. Esnault and F. Volino, Phys. Rev. Lett., 1986, 57, 1745–1748 CrossRef CAS.
- M. Lukaschek, G. Kothe, C. Schmidt, A. E. Gomes and A. Polimeno, J. Chem. Phys., 2002, 117, 4550–4556 CrossRef CAS.
- NMR of Ordered Liquids, ed. E. E. Burnell and C. E. de Lange, Kluwer, Dordrecht, 2003 Search PubMed.
- J. Li, K. G. Wilmsmeyer and L. A. Madsen, Macromolecules, 2009, 42, 255–262 CrossRef CAS.
- B. Deloche and E. T. Samulski, Macromolecules, 1981, 14, 575–581 CrossRef CAS.
- M. Stefanov and A. Saupe, Mol. Cryst. Liq. Cryst., 1984, 108, 309–316 CrossRef CAS.
- D. Vijayaraghavan and K. A. Suresh, Mol. Cryst. Liq. Cryst., 2005, 434, 609–618 CAS.
- F. C. Frank, Discuss. Faraday Soc., 1958, 25, 19–28 RSC.
- O. Parodi, J. Phys.-Paris, 1970, 31, 581–584 CAS.
- M. Miesowicz, Nature, 1946, 158, 27 CrossRef CAS.
- J. L. Ericksen, Trans. Soc. Rheol., 1961, 5, 23–34 CrossRef CAS.
- F. M. Leslie, Arch. Ration. Mech. Anal., 1968, 28, 265–283 CrossRef.
- B. S. Douglass, R. H. Colby, L. A. Madsen and P. T. Callaghan, Macromolecules, 2008, 41, 804–814 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1sm06634j |
| This journal is © The Royal Society of Chemistry 2012 |