Surface adsorption of fibronectin-derived peptide fragments: the influence of electrostatics and hydrophobicity for endothelial cells adhesion†
Cristina
Satriano
*a,
Maria Elena
Fragalà
a,
Giuseppe
Forte
b,
Anna Maria
Santoro
c,
Diego
La Mendola
c and
Bengt
Kasemo
d
aDepartment of Chemical Sciences and INSTM, Catania University, viale Andrea Doria, 6, 95125, Catania, Italy. E-mail: csatriano@unict.it; Fax: +39 095 580138; Tel: +39 095 7385136
bDepartment of Pharmaceutical Sciences, Catania University, viale Andrea Doria, 6, 95125, Catania, Italy
cInstitute of Biostructures and Bioimages, CNR-Catania, viale Andrea Doria, 6, 95125, Catania, Italy
dDepartment of Applied Physics, Chalmers University of Technology, Gothenburg, Sweden
First published on 11th November 2011
Abstract
The adsorption on hydrophobic and hydrophilic silica-based surfaces of the integrin-binding PHSRN peptide and the single-residual-mutated analogues, PHSEN and PHSFN, is investigated by comparative QCM-D, XPS, SFG measurements and molecular dynamics calculations. Endothelial cell cultures on the peptide-functionalized materials highlight their tunable pro- or anti-angiogenic potential.
The control of angiogenesis is a great challenge in therapeutic and regenerative medicine.1 Crucial for the activation of the angiogenic processes is the interaction of endothelial cells (ECs) with the extracellular matrix (ECM), which, in turn, transmits signal cascades that mediate major angiogenic responses, including EC proliferation and migration.2 One current approach to design and fabricate solid matrix able to guide ECs adhesion is the surface decoration with and/or incorporation of pro-/anti-angiogenic factors.3 Fibronectin (FN) is an adhesive glycoprotein ubiquitously expressed in ECM and body fluids, which plays a critical adhesive role during development, tissue repair and angiogenesis.4 The use of FN peptide fragments, able to mimic the protein activity, is very attractive as alternative of using the whole protein, since a proper peptide design allows triggering the specific binding between the substrate and biomolecule, and the subsequent interaction with ECs.5
The Pro-His-Ser-Arg-Asn (PHSRN) sequence is located in the contiguous domain of the Arg-Gly-Asp (RGD) epitope, which is the first FN recognition site. Recently, PHSRN has been demonstrated to act by itself as an integrin binding peptide, by enhancing ECs adhesion and proliferation.6
We have previously shown that PHSRN molecules can be immobilized both on hydrophilic and hydrophobic silica-based surfaces by irreversible adsorption; the resulting adlayers exhibit comparable peptide coverage but an enhanced fibroblast cells spreading on the hydrophilic substrate.7
The present study aims to scrutinize the role of electrostatics and hydrophobic forces for driving the peptide–surface interaction and, in turn, the ECs response to the same solid matrices surface-decorated by the adsorbed peptide fragments. The single-point-mutated peptides PHSEN and PHSFN have been synthesized in order to substitute the positively charged arginine (R) residue in PHSRN with glutamic acid (E) and phenylalanine (F), respectively. The estimated isoelectric pH values are ∼5 for PHSEN, ∼7 for PHSFN and ∼10 for PHSRN, respectively. Moreover, the phenylalanine residue induces a less hydrophilic character to the sequence through the presence of the benzene ring (Chart 1).
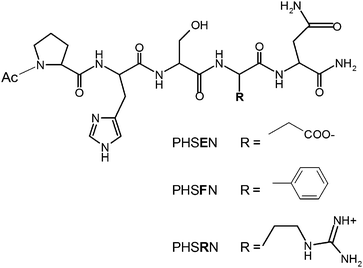 | ||
| Chart 1 | ||
The used substrates, at the pH of 7.4, consist respectively of a deprotonated silica surface, with a water contact angle of less than 10°, which will be noted as S1 and a partially methylated polysiloxane surface (O−/CH3 ratio of 1/3),8 with a water contact angle of about 90°, henceforth noted as S2.
The peptide adsorption kinetics, the coverage and average orientation on the two surfaces were investigated by quartz crystal microbalance with dissipation monitoring (QCM-D), X-ray photoelectron spectroscopy (XPS) and sum frequency generation (SFG) spectroscopy.
The QCM-D curves (Fig. 1) show that for the negatively charged PHSEN the shifts of frequency (Δf) and dissipation (ΔD) upon peptide addition are larger on the hydrophilic S1 (Fig. 1a) than on the hydrophobic S2 (Fig. 1b). Curve shifts about one order of magnitude lower, and with an opposite trend, are found for the more hydrophobic PHSFN fragment (Fig. 1c and d). As a matter of fact, the Δf values on S1 and S2 measured for the irreversible bound peptide, i.e., after the buffer rinsing step, are respectively about −50 Hz and −37 Hz for PHSEN and −1 Hz and −2 Hz for PHSFN.
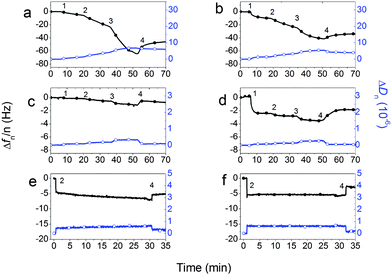 | ||
| Fig. 1 QCM-D curves of Δf (solid symbols) and ΔD (open symbols) for adsorption of PHSEN on S1 (a) and S2 (b) and of PHSFN on S1 (c) and S2 (d). Time points indicated correspond to subsequent peptide additions (1 = 1.5 mM; 2 = 3 mM; 3 = 6 mM) and buffer rinsing step (4). Adsorption curves of 3 mM PHSRN on S1 (e) and S2 (f) are shown for comparison. | ||
Moreover, ΔD curves evidence a peptide-dependent more than a substrate-dependent trend. In fact, after buffer rinsing, for both S1 and S2 substrates the ΔD is of about +5 × 10−6 for PHSEN, and of ∼+0.1 × 10−6 for PHSFN. What is more, in the results in Fig. 1a the kinetics is peculiar with an increasing rate of adsorption as coverage increases. Such kinetics suggest that the already adsorbed molecules promote the adsorption of new molecules, i.e., the already adsorbed molecules increase the sticking coefficient. It has to be mentioned that Δf provides information on the mass uptake at the sensor surface due to the peptide adsorption process (Δf ∝ Δm), while ΔD is related to the viscoelastic behavior of the peptide–substrate interface.9 In our case the relatively low changes of dissipation over frequency for PHSFN (i.e., ΔD/Δf around 0.05–0.1) point to the formation of a rigid and uniform adsorbed layer. This allows conversion of Δf to Δm, the actual adsorbed mass, which corresponds respectively to about 60% on S2 and 30% on S1 of the ideal monolayer coverage.‡ Noteworthy, similar peptide coverages were obtained for the wild type PHSRN sequence on the same S1 and S2 surfaces7 and are confirmed at the present conditions (Fig. 1e and f).
As to PHSEN, the relatively high dissipation shifts (i.e., ΔD/Δf ≈ 0.1–0.14) suggest relevant contributions of the dissipative losses for both adlayers on S1 and S2. In such cases the hydrodynamic mass calculated by Sauerbrey approximation is not accurate enough to estimate the actual mass of adsorbed peptide and comparative analysis with another surface sensitive technique is desirable. Accordingly, the in situ QCM-D peptide adsorption measurements and the related hydrated masses have been compared to ex situ XPS analyses and the related coverage of dry peptide masses.
The XPS quantitative results of average surface composition, before and after peptide immobilization (Table S1 in the ESI†), indicate comparable coverages for all three peptides both on S1 and S2, as evidenced by the appearance of nitrogen signal (about 2%) and the parallel decrease of silicon signal from the substrate. Moreover, the C 1s photoelectron spectra of PHSEN–S1 (Fig. 2a), PHSEN–S2 (Fig. 2b) and PHSRN–S2 (Fig. 2f) show the increase of the intensity ratio between the peaks related to the polar C–O bonds (at a binding energy of 286.4 ± 0.2 eV) and to the hydrocarbon C–C and C–H bonds (at 285.0 ± 0.2 eV) compared to the corresponding bare substrates (Fig. 2g and h).
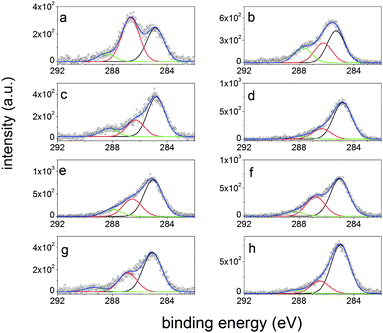 | ||
| Fig. 2 C 1s photoelectron spectra of peptide-decorated and correspondent bare surfaces: (a) PHSEN–S1; (b) PHSEN–S2; (c) PHSFN–S1; (d) PHSFN–S2; (e) PHSRN–S1; (f), PHSRN–S2; (g) S1; (h) S2. | ||
For PHSFN–S1 (Fig. 2c) the weight of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O component (at a binding energy of 288.0 ± 0.2 eV) is much higher than for PHSFN–S2 (Fig. 2d). These results give a clear indication of effective substrate coverage but also point to different average orientations for the three peptides at the interface with S1 and S2.
O component (at a binding energy of 288.0 ± 0.2 eV) is much higher than for PHSFN–S2 (Fig. 2d). These results give a clear indication of effective substrate coverage but also point to different average orientations for the three peptides at the interface with S1 and S2.
In order to get a deeper understanding of the biomolecule–substrate interface, the peptide fragment orientation on S1 and S2 was scrutinized by additional experiments (QCM-D, SFG) and molecular dynamics (MD) calculations.
Fig. 3 shows the dissipation versus frequency plots for PHSEN, PHSFN and PHSRN on S1 and S2.
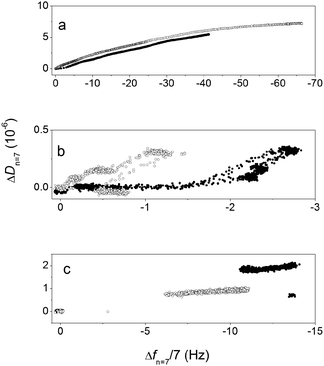 | ||
| Fig. 3 D–f plots for PHSEN (a), PHSFN (b) and PHSRN (c) adsorption on S1 (open symbols) and S2 (solid symbols). | ||
Such plots, by taking out the time as an explicit variable, give an indication of looser or stiffer molecular arrangement at the interface in relation to a higher or lower curve slope, respectively. Comparable curve slopes are found for the negatively charged PHSEN (Fig. 3a) on both S1 and S2, while for PHSFN (Fig. 3b) and PHSRN (Fig. 3c) the slopes are higher in the initial adsorption phases respectively on S1 and S2. Moreover, the adsorption process is slower for PHSEN and PHSFN than for PHSRN, the latter showing an abrupt change of frequency and dissipation at the beginning of the adsorption process, while the other two peptides exhibit a homogeneous data points dispersion for the whole process. This evidence points to the major role of attractive electrostatic forces between the positively charged PHSRN and the negatively charged S1 and S2 surfaces7 and suggests that for both PHSFN and PHSRN the substrate surface polarity may drive different peptide orientations while this is not the case for the negatively charged PHSEN.
According to QCM-D, the SFG results evidence a surface-dependent structuring of water at the solid–peptide interface for PHSFN and PHSRN but not for PHSEN. Fig. 4 shows the SFG spectra for S1 and S2 at the air–solid (Fig. 4a and b) and peptide–solid (Fig. 4c and d) interfaces. As to the bare substrates, the two spectral regions of CHx (1 < x < 3) stretching, in the 2800–3000 cm−1 range, and of –OH stretching, in the 3200–3600 cm−1 range, show different characteristic features. In fact, the hydrophobic S2 spectrum is dominated by the peaks at 2850, 2875, 2915, and 2940 cm−1, assigned respectively to the symmetric CH2, CH3, C–H, Si–CH3 stretching modes and to CH3 Fermi resonance modes.10,11 Conversely, the hydrophilic S1 surface shows a much lower contribution of all these C–H components at the used experimental conditions (see ESI†).
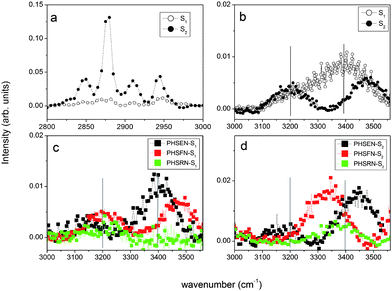 | ||
| Fig. 4 SFG spectra for: (a) CHx stretch region for S1 and S2; (b) OH stretching modes for S1 and S2; OH stretching modes for peptides–S1 (c) and peptides–S2 (d). | ||
As to the peptide–S1(2) interfaces, no clear C–H vibrational modes were observed, while a measurable influence from interfacial water as a function of peptide identity is found in the –OH stretching region. Such a region is characterized by a peak at ∼3200 cm−1, attributed primarily to “ice-like water”, i.e., coupled OH symmetric stretchings of tetrahedrally coordinated water molecules, and one peak at 3400 cm−1, named “liquid-like water”, due to the disordered H-bonds network of asymmetrically bound water molecules.12–14 For PHSRN–S1 and PHSFN–S1 interfaces (Fig. 4c), the peak at ∼3400 cm−1 completely disappears in the spectrum, while PHSRN–S2 and PHSFN–S2 show a predominant broad component at ∼3300 cm−1, likely due to the N–H stretch (Fig. 4d).15 Interestingly, for both PHSEN–S1 and PHSEN–S2 similar spectral features are found, with the reduction of the component at ∼3200 cm−1 and the enhancement of that at ∼3400 cm−1. These findings indicate that the adsorbed peptides influence the ordering of water molecules at the interface with the solid, depending on the specific matching of attractive electrostatic and/or hydrophobic forces at the interface.
The MD calculations support the experimental evidence showing: (i) that the interfacial water layer between the peptide molecules and the substrate surface has a thickness depending on the substrate more than the peptide nature; (ii) remarkable differences in the orientation of carbonyl, aromatic ring and charged groups for the three peptides at the interface with S1 or S2.
As to the interfacial water layer between the substrate and the peptide molecules, the thickness values of about 0.85 nm for S1 and of 0.25 nm for S2 were calculated for all three cases (Fig. S1 in the ESI†). Moreover, the statistically significant molecular configurations worked out during 2 ns MD simulation (Fig. 5) show that the oxygen atoms of the peptide backbone carbonyl groups are turned away from S1, likely because of electrostatic repulsion with the negative SiO− groups on the surface, while they have a random orientation towards S2. Such an arrangement is more pronounced for PHSRN and PHSFN. All three peptides exhibit an orientation of the histidine imidazole ring (Fig. 5, green shadow) either parallel to the hydrophobic S2 or orthogonal to the hydrophilic S1 surface, due to weak van der Waals forces. The time evolution analysis of imidazolic and (for PHSFN) phenyl ring dihedral angles supports this observation (Fig. S2, in the ESI†). Finally, the serine side chain –OH group (Fig. 5, magenta shadow) generally points towards the bulk solution and far from the surface, the only exception being the case of PHSRN–S1, where the stronger electrostatic interactions due to the polar and positively charged guanidine group (Fig. 5c, blue shadow) are likely to drive the hydroxyl orientation.
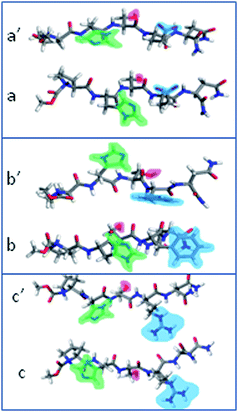 | ||
| Fig. 5 Final geometries obtained by simulation of: PHSEN on S1 (a) and S2 (a′), PHSFN on S1 (b) and S2 (b′), PHSRN on S1 (c) and S2 (c′). | ||
Proof-of-working experiments of pro- or anti-angiogenic effects to the peptide adlayers on S1 and S2 were performed by short incubation (3 h) of HUVEC cells in essential medium, i.e., in the absence of nutrients such as growth factors and serum, followed by 22 h of incubation in the medium supplemented with nutrients. The starved conditions at the beginning of cell–surface interaction are requested to reduce as much as possible any interference with the components of the culture media.
Fig. 6 shows that PHSEN–S1 and PHSRN–S1 are better adhesive substrates than the control and good mimic candidates for the whole FN, while the viable cells on PHSFN–S1 surfaces are significantly reduced in comparison with both the bare S1 as well as the PHSEN- and PHSRN-immobilized layers. On the other hand, all three peptide–S2 substrates as well as FN–S2 exhibit an almost comparable response than the bare control S2 surface. These results highlight the good potentiality of the charged and the hydrophobic peptides to act respectively as pro- or anti-angiogenic factors on the hydrophilic S1 substrate.
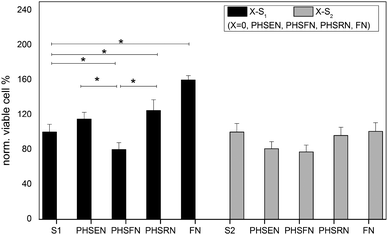 | ||
| Fig. 6 Viability data from MTT assay of HUVEC adhered on the peptide–S1 (black) and peptide–S2 (gray). Data are normalized to the correspondent bare substrate S1 (or S2). The FN–S1(2) substrates were prepared by 20 min adsorption of 10 μM fibronectin solution in PBS. Data from experiments in triplicate (* p < 0.01, two-tailed Anova test). | ||
In conclusion, this comparative experimental and theoretical investigation evidences that the replacement of the arginine residue of PHSRN with either glutamic acid (PHSEN) or phenylalanine (PHSFN) residues significantly affects the charge interactions between the oligopeptides and the two investigated substrates, respectively a hydrophilic deprotonated silica surface and a hydrophobic partially methylated polysiloxane surface. The resulting biomolecule-decorated surfaces, after the irreversible peptide adsorption process, are very promising for triggering endothelial cells adhesion. Further investigations will be aimed to the fine tuning of the peptide immobilization process, by changing both the length and composition of the primary sequence, as well as to an in-depth study of the cell response, in terms of cell proliferation, differentiation, migration, and the integrin signalling pathways involved in angiogenic processes.
Notes and references
- E. A. Phelps and A. J. Garcia, Regener. Med., 2009, 4, 65 CrossRef.
- H. M. Eilken and R. H. Adams, Curr. Opin. Cell Biol., 2010, 22, 617 CrossRef CAS.
- M. Lovett, K. Lee, A. Edwards and D. L. Kaplan, Tissue Eng., Part B: Rev., 2009, 15, 353 CrossRef CAS.
- R. Pankov and K. M. Yamada, J. Cell Sci., 2002, 115, 3861 CrossRef CAS.
- H. Cui, M. J. Webber and S. I. Stupp, Biopolymers, 2010, 94, 1 CrossRef CAS.
- A. Hattori, K. Hozumi, J. A. Ko, T. Chikama, K. Oomikawa, J. Kato, K. Ishida, N. Hoshi, F. Katagiri, Y. Kikkawa, M. Nomizu and T. Nishida, Biochem. Biophys. Res. Commun., 2009, 379, 346 CrossRef CAS; Y. Liu, W. Wang, J. Wang, Z. Yuan, S. Tang, M. Liu and H. Tang, Colloids Surf., B, 2011, 84, 6 CrossRef.
- C. Satriano, G. M. L. Messina, C. S. Marino, I. Aiello, E. Conte, D. La Mendola, D. A. Distefano, F. D'Alessandro, G. Pappalardo and G. Impellizzeri, J. Colloid Interface Sci., 2010, 341, 232 CrossRef CAS.
- C. Satriano, G. Marletta and B. Kasemo, Surf. Interface Anal., 2008, 40, 649 CrossRef CAS.
- E. M. Larsson, M. E. Edvardsson, C. Langhammer, I. Zorić and B. Kasemo, Rev. Sci. Instrum., 2009, 80, 125105 CrossRef.
- Y. Liu, L. K. Wolf and M. C. Messmer, Langmuir, 2001, 17, 4329 CrossRef CAS.
- B. C. Chow, T. T. Ehler and T. E. Furtak, Appl. Phys. B: Lasers Opt., 2002, 74, 395 CrossRef CAS.
- Q. Du, E. Freysz and Y. R. Shen, Science, 1994, 264, 826 CAS.
- F. Scatena, M. G. Brown and G. L. Richmond, Science, 2001, 292, 908 CrossRef.
- Q. Du, E. Freysz and Y. R. Shen, Phys. Rev. Lett., 1994, 72, 238 CrossRef CAS.
- L. Fu, J. Liu and E. C. Y. Yan, J. Am. Chem. Soc., 2011, 133, 8094 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: (1) Lengthy experimental details; (2) XPS table of average surface atomic composition for bare hydrophilic S1 and hydrophobic S2 surfaces and the corresponding peptide adlayers; (3) Fig. S1: disposition of PHSRN on S1 and S2 after 2 ns of MD simulation; and (4) Fig. S2: phenyl and imidazolic rings orientation with respect to S1 and S2 surfaces; time evolution of dihedral angle. See DOI: 10.1039/c1sm06655b |
| ‡ According to the calculated PHSFN molecular mass (641.68 g mol−1) and the 2D projected peptide dimensions (19.7 × 9.0 Å2), the ideal ‘close-packed’ monolayer coverage is estimated to be around 90 pmol cm−2. |
| This journal is © The Royal Society of Chemistry 2012 |