Edible supramolecular chiral nanostructures by self-assembly of an amphiphilic phytosterol conjugate†
Antoni
Sánchez-Ferrer
,
Jozef
Adamcik
and
Raffaele
Mezzenga
*
ETH Zurich, Food & Soft Materials Science, Institute of Food, Nutrition & Health, Schmelzbergstrasse 9, LFO, E23, 8092, Zürich, Switzerland. E-mail: raffaele.mezzenga@agrl.ethz.ch
First published on 28th October 2011
Abstract
We study the self-assembly of a food-grade glucose-β-sitosterol conjugate in bulk and in solution by small angle X-ray scattering (SAXS) and atomic force microscopy (AFM). The amphiphilic behavior of the glucose-β-sitosterol conjugate, combined with the chirality of both its moieties, leads in solutions to the aggregation into supramolecular chiral aggregates with plate-like, spherical and helical ribbon configurations, depending on concentration and solvent quality. Owing to the crucial role of phytosterols in replacing cholesterol content and the need for functional edible fibrils with enhanced nutritional value, this system shows great promise in the struggle for the design of new functional food building blocks with targeted properties.
Introduction
Phytosterols are sterols extracted from plants, and have structures similar to that of cholesterol.1–5 They are building blocks of biomembranes and precursors for hormones in plants.6,7 They have been proposed as a natural source for the reduction of both cholesterol in blood plasma as well as cholesterol low-density lipoprotein, and recommended as a dietary supplement.8–10 Besides regular phytosterols, conjugated phytosterols are also present in plants as fatty acids, acylated glycosides or glycoside derivatives.1–3,6,7,11Phytosterols solubility is very low in water,12 and their bioavailability is reduced when they are in the crystalline form.13,14 A route to make them more soluble is to conjugate them to a polar group, such as sugars, which might enhance the solubility due to the presence of hydroxyl groups.15 Alternatively, stable phytosterol colloidal particles have been obtained using non-ionic surfactants,16,17 and different sizes (from 80 to 250 nm in diameter, and from 500 to 700 nm in length) or morphologies (rod-like and plate-like crystals)18,19 have been observed.
Cholic acid ester derivatives with hydrophilic ethylene glycol tails20 and other bile acid conjugates21–24 have been reported to form gels. The former gels broke down to crystals with helical structures (pitch: 7.3 Å), with different dimensions depending on the handedness of the self-assembled structure (left-handed helical crystal: 1.6 μm diameter and 8.9 μm length; right-handed helical crystal: 4.8 μm diameter and 190 μm length). In hydrophobic media such as natural oils on the other hand, mixtures of γ-oryzanol with phytosterols associate in a synergistic way to form hollow tubular structures with diameter in the range of 6.7–8.0 nm and a wall thickness of 0.8–1.2 nm.25–29
Despite these encouraging works, the supramolecular self-aggregation behavior of these systems remains a complex, still poorly understood process, in which hydrophobic interactions, hydrogen bonding, crystallization, steric effects, and supramolecular chirality all concur to establish the final complex architectures.
In this work, we discuss for the first time the spontaneous self-assembly behaviour of a conjugated glucose–sitosterol (β-sitosterolin), synthesized in our laboratory, and an analogue of that found in almonds, cashews, sesame seeds, sunflower seeds, squash, barley, peas, olive oil, peanuts and cloves. We rely on small and wide angle X-ray scattering (SAXS, WAXS) and on atomic force microscopy (AFM). We discuss the complex architectures of the colloidal spheres, platelets and fibrils based on supramolecular β-sitosterolin self-assembly and we touch on the control of their morphologies by changing the solvent quality, concentration and aggregation time.
Experimental section
Materials
2,3,4,6-Tetra-O-acetyl-α-D-glucopyranosyl bromide (Fluka) and β-sitosterol (Sigma, ≥70%, with campesterol and β-sitostanol residual content) were used as received. Zinc bromide (Sigma-Aldrich) and the powdered 4A molecular sieves were dried at 120 °C for 24 h prior to use. CH2Cl2 was dried over CaH2 and distilled before use. Dimethyl sulfoxide (DMSO), isopropanol (iPrOH) and Milli-Q water were filtered through a PTFE syringe filter (0.2 μm) for the preparation of AFM and SAXS samples.Apparatus and techniques
1H NMR experiments were carried out at room temperature on a Bruker Avance Spectrometer operating at 400 MHz, and using CDCl3 or DMSO-d6 as solvents and as the internal standards.Simultaneous small and wide-angle X-ray scattering (SAXS and WAXS, respectively) experiments were performed using a Rigaku MicroMax-002+ microfocused beam (4 kW, 45 kV, 0.88 mA) in order to obtain direct information on the SAXS and WAXS reflections. The Cu Kα radiation (λCu Kα = 1.5418 Å) was collimated by three pinhole (0.4, 0.3, and 0.8 mm) collimators. The scattered X-ray intensity was detected by a Fuji Film BAS-MS 2025 imaging plate system (15.2 × 15.2 cm2, 50 mm resolution) and a two-dimensional Triton-200 X-ray detector (20 cm diameter, 200 mm resolution), for WAXS and SAXS regions, respectively. An effective scattering vector range of 0.05 nm−1 < q < 25 nm−1 was obtained, where q is the scattering wave vector defined as q = 4πsin θ/λCu Kα, with a scattering angle of 2θ.
Fourier-transform infrared (FTIR) spectra of solid samples were recorded at room temperature with a Varian 640 FTIR spectrometer and a MKII Golden Gate single attenuated total reflection (ATR) system.
ORD experiments were performed on an ORDM Jasco-815 polarimeter with a 5 cm length and 1 cm diameter cuvette in DMSO as solvent at 25 °C, and for the observation of the change in the optical rotation as function of the wavelength from 300 to 700 nm.
Tapping mode AFM was carried out on a Nanoscope 8 Multimode Scanning Force Microscope (Veeco). AFM cantilevers (Veeco, USA) for tapping mode in soft tapping conditions were used at a vibrating frequency of 150 kHz. Images were simply flattened using the Nanoscope 8.1 software, and no further image processing was carried out. A 30 μL aliquot of solution at a certain concentration of β-sitosterolin was deposited onto freshly cleaved mica or on highly ordered pyrolytic graphite (HOPG), incubated for 1 minute and dried in air.
Synthesis of β-sitosterolin tetraacetate (β-sitosteryl-2,3,4,6-tetra-O-acetyl-β-D-glucopyranoside)
In a 100 mL round-bottomed flask, 1.000 g (2.43 mmol) of 2,3,4,6-tetra-O-acetyl-α-D-glucopyranosyl bromide, 1.513 g (3.65 mmol) of β-sitosterol, 1.643 g (7.30 mmol) of zinc bromide, and 2.432 g of powdered 4A molecular sieves were placed, and after purging with nitrogen, 40 mL of anhydrous CH2Cl2 were added. The suspension was magnetically stirred and brought to reflux for 6 h under nitrogen atmosphere. The solution was filtered, washed several times with CH2Cl2 and evaporated. The residue was chromatographed in a silica gel column (EtAcO/cyclohexane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4)). Yield: 1.403 g (77%).
:4)). Yield: 1.403 g (77%).
1H NMR (400 MHz, CDCl3): δ = 5.35 (1H, m, H6), 5.20 (1H, t, H3′, J = 9.5 Hz), 5.07 (1H, t, H4′, J = 9.6 Hz), 4.95 (1H, dd, H2′, J = 9.6 Hz, J = 8.1 Hz), 4.58 (1H, d, H1′, J = 10.4 Hz), 4.25 (1H, dd, H6′, J = 12.3 Hz, J = 4.7 Hz), 4.11 (1H, dd, H6′, J = 12.2 Hz, J = 2.5 Hz), 3.67 (1H, ddd, H5′, J = 9.9 Hz, J = 4.8 Hz, J = 2.4 Hz), 3.48 (1H, m, H3), 2.35 (1H, m, H4), 2.22 (1H, m, H4), 2.07 (3H, s, CH3CO), 2.04 (3H, s, CH3CO), 2.01 (3H, s, CH3CO), 2.00 (3H, s, CH3CO), 2.00–1.00 (27H), 0.98 (3H, s, H19), 0.91 (3H, d, H21, J = 6.0 Hz), 0.84 (3H, t, H29, J = 6.3 Hz), 0.82 (3H, d, H27, J = 6.1 Hz), 0.79 (3H, d, H26, J = 6.3 Hz), 0.67 (3H, s, H18) ppm (see ESI, Chart S1†).
Synthesis of β-sitosterolin (β-sitosteryl-β-D-glucopyranoside)
In a 250 mL round-bottomed flask with magnetic stirring, 1.000 g (1.34 mmol) of β-sitosteryl-2,3,4,6-tetra-O-acetyl-β-D-glucopyranoside was dissolved in 100 mL of a mixture of 0.25 M NaOH/THF/MeOH (1:2:1). The system was magnetically stirred at room temperature and the hydrolysis was kept for 4 h. The white solid that appeared was filtered out, washed several times with a cold mixture of THF/H2O, and dried under vacuum. Yield: 0.504 g (36%).
1H NMR (400 MHz, DMSO-d6): δ = 5.33 (1H, m, H6), 4.87 (3H, s, OH), 4.42 (1H, s, OH), 4.22 (1H, d, H1′, J = 7.6 Hz), 3.63 (1H, dd, H3′, J = 10.8 Hz, J = 4.3 Hz), 3.43 (1H, m, H3), 3.18–2.96 (4H, m, H2′-H5′-2H6′), 2.89 (1H, t, H4′, J = 8.2 Hz), 2.35 (1H, m, H4), 2.12 (1H, m, H4), 2.00–1.00 (27H), 0.96 (3H, s, H19), 0.90 (3H, d, H21, J = 6.4 Hz), 0.89 (3H, t, H29, J = 6.0 Hz), 0.82 (3H, d, H27, J = 6.1 Hz), 0.80 (3H, d, H26, J = 5.9 Hz), 0.65 (3H, s, H18) ppm (see ESI, Chart S2†).
Preparation of the self-assembled samples
AFM and SAXS samples were prepared from DMSO solutions of β-sitosterolin. From an initial concentration of β-sitosterolin in DMSO, the sample was diluted 10 times by adding drop wise water or isopropanol. AFM samples had a concentration ranging from 0.1 to 0.01 w/w% of β-sitosterolin. SAXS samples had a concentration ranging from 1 to 0.1 w/w%. For the SWAXS powder diffraction experiment, β-sitosterolin was prepared by drying under vacuum the sample that appeared after the final synthetic step.Results and discussion
Synthesis and characterization of β-sitosterolin
The β-sitosterol conjugate was synthesized in two steps (Scheme 1). The first reaction was the nucleophilic substitution in which a tetraacetyl protected α-D-glucose containing bromide as a living group in the anomeric carbon reacted with the hydroxyl group from the β-sitosterol in the presence of a Lewis acid in anhydrous media.30 The final amphiphilic molecule β-sitosterolin was obtained viahydrolysis in basic conditions of the four acetyl protecting groups in the sugar moiety.31 The chemical structures of both molecules (β-sitosterolin tetraacetate and β-sitosterolin) were confirmed by their corresponding 1H NMR spectra,32–34 solubility tests, Fourier-transform infrared spectroscopy (FTIR) and optical rotatory dispersion (ORD) experiments. | ||
| Scheme 1 Synthetic route followed to produce β-sitosterolin. | ||
D-Glucose (δD-glucose = 37.8 MPa1/2, log PD-glucose = −1.88)35 is soluble in DMSO (δDMSO = 26.7 MPa1/2, log PDMSO = −1.35)36,37 and water (δwater = 47.8 MPa1/2),36,37 but insoluble in iPrOH (δiso-PrOH = 23.5 MPa1/2, log Piso-PrOH = 0.16).36,37 β-Sitosterol (δβ-sitosterol = 18.4 MPa1/2, log Pβ-sitosterol = 10.73)38 is soluble in DMSO and iPrOH, but not in water. The amphiphilic molecule β-sitosterolin (δβ-sitosterolin = 28.1 MPa1/2, log Pβ-sitosterolin = 8.78) is soluble only in DMSO, and when adding water or iPrOH a precipitate, which remains stable as a suspension, is formed. Thus, DMSO is a good solvent for both moieties in β-sitosterolin, and water and iPrOH are only selective solvents for one of the building blocks. As it will be shown below, this enables for the control of the self-aggregating behaviour of β-sitosterolin by dispersing its DMSO molecular solution into either of the two selective solvents.
ORD experiments show the optical activity of the conjugated molecule in DMSO, when the molecule is fully soluble. D-Glucose is a dextrorotatory molecule with a specific rotation of [α]D = +57.3° measured at 25 °C in DMSO, while both β-sitosterol and β-sitosterolin are levorotatory molecules with specific rotations of [α]D = −18.6° and [α]D = −10.0°, respectively (Fig. 1a). The low value of the specific rotation of β-sitosterolin39 is due to the presence of co-existing β-sitostanolin, which is a dextrorotatory molecule and averages the final optical rotatory power of the phytosterol conjugate.
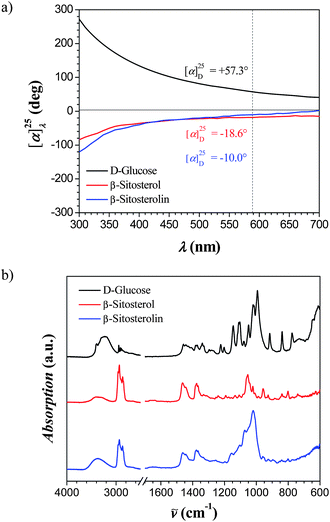 | ||
| Fig. 1 (a) Optical rotatory dispersion (ORD) spectra of D-glucose, β-sitosterol and β-sitosterolin as function of the transmitted wavelength. (b) ATR-FTIR spectra of D-glucose, β-sitosterol and β-sitosterolin showing the presence of characteristic vibrational modes from both D-glucose and β-sitosterol. | ||
ATR-FTIR experiments of β-sitosterolin showed the presence of characteristic vibrational modes from both D-glucose (st OH, st, C–OH, and st C–O–C) and β-sitosterol (st C–H, st and δ C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, and δ from CH3 and CH2) (Fig. 1b).
C, and δ from CH3 and CH2) (Fig. 1b).
Self-assembly of β-sitosterolin
In order to understand the driving force to form supramolecular aggregates in a selective solvent, we first studied the self-assembly behaviour in bulk, where the head–tail interactions of β-sitosterolin are not mediated by either iPrOH or water. A very strong micro-phase separation between the polar glucose head group and the apolar rigid sitosteryl tail is found, as revealed by the Small and Wide Angle X-ray Scattering (SWAXS) diffractogram of the dry powder sample shown in Fig. 2 after drying the synthesized sample under vacuum.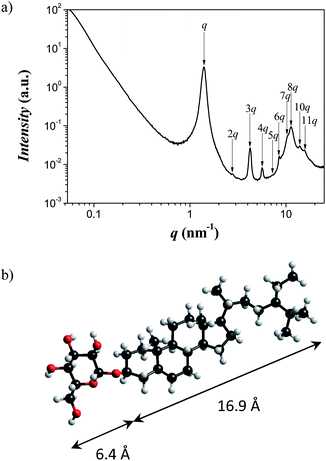 | ||
| Fig. 2 (a) SWAXS profile pattern for β-sitosterolin in the solid state indicating self-assembly into a lamellar phase of period 4.5 nm. (b) The chemical structure and dimensions of the polar head and apolar tail group, match precisely the half-period of the lamellar phase. | ||
The eleven Bragg diffraction peaks with a spacing of d = 4.5 nm and a correlation length of ξ = 49.3 nm indicate a perfectly ordered lamellar phase, in which the β-sitosterolin molecules form bilayered domains composed of two facing glucose heads and two sitosteryl skeletons (Fig. 2), and witness the very high incompatibility between the polar head and hydrophobic tail of β-sitosterolin.
When adding drop-wise water or iPrOH (poor solvents) into a DMSO (good solvent) solution of β-sitosterolin, cloudy dispersions were rapidly obtained (see ESI, Fig. S1†). The SAXS pattern of these dispersions showed the presence of different types of aggregates (Fig. 3). β-Sitosterolin was also dispersed in pure iPrOH which was stable upon time, while once dispersed in pure water it precipitated. This is the basis for understanding the relatively flat signal of 1 wt% β-sitosterolin in H2O shown in Fig. 3a, as opposed to all other scattering profiles.
 | ||
| Fig. 3
SAXS pattern for dispersions of (a) 1 w/w% β-sitosterolin in pure H2O, 1 w/w% in H2O/DMSO (9:1), 0.1 w/w% in H2O/DMSO (9:1), and (b) 1 w/w% β-sitosterolin in pure iPrOH, 1 w/w% in iPrOH/DMSO (9:1), 0.1 w/w% in iPrOH/DMSO (9:1) together with their corresponding fitting curves. Note: the continuous top black curve is the scattering pattern of solid β-sitosterolin. | ||
All samples prepared by addition of iPrOH or water in DMSO show a scattering peak at around q = 1.40 nm−1 (arrows in Fig. 3) which corresponds to the micro-phase separation length scale 4.5 nm already observed in the solid state. Thus, micro-phase separation at this small length scale is maintained also in solution, irrespective of the selective solvent considered. However, this peak was considerably sharper in the case of water as solvent.
From the shape of the peak at q = 1.40 nm−1, the correlation length ξ was evaluated and by diluting the sample a decrease in its value was observed. Samples in iPrOH/DMSO (9:1) (Fig. 3b) showed correlation lengths from 16.4 to 3.6 nm at 1 and 0.1 w/w% concentrations, respectively. For samples in H2O/DMSO (9:1), values from 35.1 to 8.2 nm were found at the same respective concentrations, indicating that water promotes more strongly correlated structures compared to iPrOH, probably due to the high insolubility (stronger aggregation) of the β-sitosterolin molecule in water (see ESI, Fig. S2†).
Another key point is the shape of scattering curves in the low q limit (0.06–1.0 nm−1). In water, a q−2 power-law decrease is observed, indicative of platelet-like sharp interfaces, while in iPrOH, samples show a bump with a slope value from 3.5 to 3.8, indicative of polydisperse sphere-like structures, with average radius from 20 to 30 nm. This is an indication of different morphological structures in the aggregates at larger length scales ranging from 6 to 100 nm (see ESI, Fig. S3†).
A first rationale into the shape of the expected morphologies in either water or iPrOH can be obtained by considering the mismatch in volume fraction of the polar head group (ϕGlucose = 0.20) and the rigid apolar tail (ϕSitosterol = 0.80): when iPrOH is present, β-sitosterol will be exposed against the solvent and ϕSitosterol/ϕGlucose = 4.1 is high enough to enable the formation of spherical-like micelles. On the other hand, when water makes up the majority of the solvent, glucose will be exposed against water, and the ratio ϕGlucose/ϕSitosterol = 0.25 is so low that spherical micelles are no longer expected and aggregates with a more flat-like interface will be promoted. The fact that the q = 1.40 nm−1 reflection is maintained both in the solid state and in solvent dispersions, that is to say that no swelling of the bilayer spacing is observed, further suggests that solvation effects should not greatly affect the ϕGlucose/ϕSitosterol and the reasoning given above.
Further insight into the structures of the aggregates was gained by AFM. At high concentrations (1 w/w%), samples in H2O/DMSO (9:1) were confirmed to adopt a flat-like structure—glucose occupying the outer part of the flat side (see ESI, Fig. S4†)—in which the thickness is clearly defined by multi-layers of steps of 4.7 nm (Fig. 4a), so that the total width of these flat-like structures is an integer multiple of 4.7 nm. At lower concentrations (0.1 w/w%), some flat-like objects break down into highly elongated ribbons wrapping into helical ribbons of typical diameter of 24 ± 5 nm and pitch of 90 ± 10 nm. Fig. 4b gives a typical AFM image for these helical ribbons, while the height profile allows unambiguous identification of the topology of the ribbon as helical. The transition from flat-like structure to helical ribbons can be explained by the diminishing probability to form large plates when decreasing the concentration. Indeed, helical ribbons are known to undergo structural transitions associated with their width.40 The real width of the ribbon W was estimated using the equation W = Ssin γ, where γ is the tilt angle of the helical ribbon edges with respect to the fibril axis (Fig. 4b). By measuring the average width of helical ribbons S = 60 ± 10 nm in the full-width half-maxima of the cross-section and measuring a tilt angle of γ = 40° ± 5°, one easily finds a typical width of 39 ± 5 nm for helical ribbons. These ribbons can then coil up due to the chirality of the rigid sitosterol skeleton molecule, while maintaining the sugar moieties exposed against the water-rich solvent. We stress here that due to the low volume fraction of helical ribbons vs.platelets and the consistency of the large width of the ribbons with the q−2 slope measured at q < 0.2 nm−1, SAXS analysis alone would not allow resolution of these helical ribbons. The handedness of the ribbons, identified as left as in the case of the sitosterol unit, demonstrates that supramolecular chirality assists the coiling of the ribbons. Previous circular polarized light experiments showed a selective reflection at certain frequency for which the left polarized light matched the same handedness as the steroidal medium.41,42
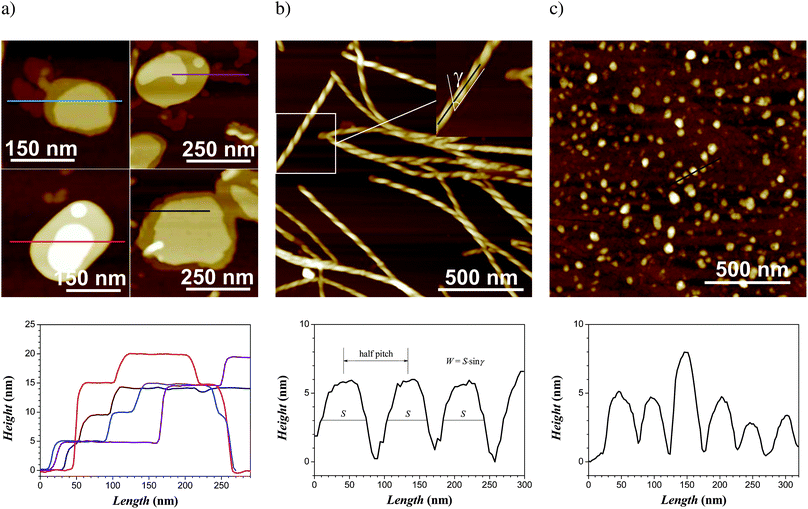 | ||
| Fig. 4
AFM height images of the self-assembly of 0.1 w/w% β-sitosterolin in H2O/DMSO (9:1) showing (a) platelet-like structures and their height profile with quantized thickness and (b) coexisting helical ribbons with the corresponding analysis of the height profile and the pitch. AFM height images of the self-assembly of 0.1 w/w% β-sitosterolin in iPrOH/DMSO (9:1) showing (c) spherical aggregates and their height profile. From (a) to (c), the lower row gives the height profile measured along the colored lines of the corresponding upper AFM images. | ||
Samples in iPrOH showed rarely the population of plate-like structures (sitosterol exposed against the solvent in the outer part of the plates) of steps of 4.7 nm, similar to water, and the dominant population was that of objects of 4 to 5 nm in height and 40–50 nm in diameter (Fig. 4c), but without quantized heights. This immediately indicates that these aggregates are larger than single β-sitosterolin micelles. The SAXS analysis shown in Fig. 3b suggests spherical aggregates of 20–30 nm radius, with an internal feature size corresponding to 2π/q, with q = 1.4 nm−1 (e.g. 4.5 nm); while the diameter observed by AFM is in the range of features extracted from the SAXS analysis, the lower height observed by AFM for these objects, which is consistent with a bilayer or a single micelle, could result from the spreading of a multi-micellar aggregate into the surface of the substrate upon adsorption. As already discussed, the high ratio in the sitosterol/glucose volume fraction does not promote in the present case the formation of self-organized aggregates with large lateral feature size, stabilizing spherical-like aggregates with smaller size.
The formation of the supramolecular colloidal aggregates reported here is caused by the sudden change in the solvent quality (polarity, hydrogen bonding, apolar interactions) of the solution. Addition of a co-solvent43–47 which is miscible with the good initial organic solvent but which is selective for specific moieties of β-sitosterolin induces the self-assembly of this molecule into supramolecular aggregates as schematically summarized in Fig. 5. The final, self-assembled structures observed here bear several similarities with the topologies of other supramolecular aggregates based on model peptides40,48 and surfactants49,50 inferring the generality of the self-assembly mechanisms involved. Although several research reports have been published for colloidal particles formed from chiral molecules,51–59 the relation between molecular chirality and supramolecular chirality is not conclusively solved yet, and the present findings can provide additional elements to the process of unravelling this complex and fascinating problem.
 | ||
| Fig. 5 Self-assembly of β-sitosterolin: (a) bilayer aggregation, (b) lamination and helical winding of the bilayer aggregates in the presence of water, and (c) spherical aggregation in the presence of isopropanol. | ||
Conclusions
The self-assembly of a phytosterol conjugate (β-sitosterolin) has been investigated in bulk, in molecular solution and in the presence of a selective solvent. Depending on the quality of the selective solvent, the molecule aggregates into molecular bilayers, in which the moieties facing the solvent can be either the glucose or sitosterol units, depending on whether water or isopropanol is used as solvent, respectively.In the first case (water), platelet-like structures with thickness equal to an integer multiple of the bilayer period (4.7 nm) are formed, which upon dilution transform into left-handed helical ribbons, whose supramolecular chirality is induced by packing of the sitosterol moieties. In the second case (isopropanol), spherical-like objects much smaller in size are formed, as a consequence of the high volume fraction of the sitosterol moieties as opposed to that of glucose.
These findings may both contribute to the understanding of supramolecular chirality in the self-aggregation process of chiral molecules, as well as to provide an appealing new system for the design of edible fibrils with desirable nutritional components.
Acknowledgements
The authors acknowledge Prof. Laura Nyström (ETH Zurich) for valuable and inspiring discussions.References
- L. J. Goad, Methods Plant Biochem., 1991, 7, 369–434 Search PubMed.
- L. J. Goad and T. Akihisa, Analysis of Sterols, Blackie, London, 1997 Search PubMed.
- N. V. Kovganko and Z. N. Kashkan, Chem. Nat. Compd., 1999, 35, 479–497 CrossRef CAS.
- R. E. Ostlund, Curr. Opin. Lipidol., 2004, 15, 37–41 CrossRef CAS.
- R. E. Ostlund, Lipids, 2007, 42, 41–45 CrossRef CAS.
- A. Tietz, Z. Naturforsch., C: J. Biosci., 1981, 36, 900–901 Search PubMed.
- M. A. Hartmann and P. Benveniste, Methods Enzymol., 1987, 148, 632–650 Search PubMed.
- A. Ambring, P. Friberg, M. Axelsen, M. Laffrezen, M. R. Taskinen, S. Basu and M. Johansson, Clin. Sci., 2004, 106, 519–525 Search PubMed.
- S. M. Mel'nikov, J. W. M. S. ten Hoorn and A. P. A. M. Eijkelenboom, Chem. Phys. Lipids, 2004, 127, 121–141 CrossRef CAS.
- L. Calpe-Berdiel, J. C. Escolà-Gil and F. Blanco-Vaca, Atherosclerosis, 2009, 203, 18–31 CrossRef CAS.
- M. Ohnishi and Y. Fujino, Phytochemistry, 1981, 20, 1357–1358 CrossRef CAS.
- G. Brufau, M. A. Canela and M. Rafecas, Nutr. Res., 2008, 28, 217–225 CrossRef CAS.
- O. J. Pollak, Circulation, 1953, 7, 702–706 CAS.
- V. Piironen, D. G. Lindsay, T. A. Miettinen, J. Toivo and A. M. Lampi, J. Sci. Food Agric., 2000, 80, 939–966 CrossRef CAS.
- P. Breinhölder, L. Mosca and W. Lindner, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2002, 777, 67–82 CrossRef.
- L. Rossi, J. W. M. Seijen ten Hoorn, S. M. Melnikov and K. P. Velikov, Soft Matter, 2010, 6, 928–936 RSC.
- L. Rossi, S. Sacanna and K. P. Velikov, Soft Matter, 2011, 7, 64–67 RSC.
- L. I. Christiansen, J. T. Rantanen, A. K. von Bonsdorff, M. A. Karjalainen and J. K. Yliruusi, Eur. J. Pharm. Sci., 2002, 15, 261–269 CrossRef CAS.
- H. Kawachi, R. Tanaka, M. Hirano, K. Igarashi and H. Ooshima, J. Chem. Eng. Jpn., 2006, 39, 869–875 CrossRef CAS.
- X. Wang, Y. Lu, Y. Duan, L. Meng and C. Li, Adv. Mater., 2008, 20, 462–465 CrossRef CAS.
- S. Mukhopadhyay and U. Maitra, Curr. Sci., 2004, 87, 1666–1683.
- P. Babu, N. M. Sangeetha and U. Maitra, Macromol. Symp., 2006, 241, 60–67 CrossRef CAS.
- S. Banerjee, R. K. Das and U. Maitra, J. Mater. Chem., 2009, 19, 6649–6687 RSC.
- P. Terech, S. Dourdain, S. Bhat and U. Maitra, J. Phys. Chem. B, 2009, 113, 8252–8267 CrossRef CAS.
- M. Pernetti, K. F. van Malssen, E. Flöter and A. Bot, Curr. Opin. Colloid Interface Sci., 2007, 12, 221–231 CrossRef CAS.
- A. Bot, R. den Adel and E. C. Roijers, J. Am. Oil Chem. Soc., 2008, 85, 1127–1134 CrossRef CAS.
- A. Bot, Y. S. J. Veldhuizen, R. den Adel and E. C. Roijers, Food Hydrocolloids, 2009, 23, 1184–1189 CrossRef CAS.
- A. Bot, R. den Adel, E. C. Roijers and C. Regkos, Food Biophys., 2009, 4, 266–272 CrossRef.
- M. A. Rogers, Food Res. Int., 2009, 42, 747–753 CrossRef CAS.
- T. Murakami, Y. Sato and M. Shibakami, Carbohydr. Res., 2008, 343, 1297–1308 CrossRef CAS.
- C. Gauthier, J. Legault, M. Lebrun, P. Dufour and A. Pichette, Bioorg. Med. Chem., 2006, 14, 6713–6725 CrossRef CAS.
- J. Seki, A. Okita, M. Watanabe, T. Nakagawa and K. Honda, J. Pharm. Sci., 1985, 74, 1259–1264 CrossRef CAS.
- E. Isik, T. Sabudak and S. Oksuz, Chem. Nat. Compd., 2007, 43, 614–615 CrossRef CAS.
- O. Akpinar and M. H. Penner, J. Carbohydr. Chem., 2008, 27, 188–199 CrossRef CAS.
- K. L. Hoy, J. Paint Technol., 1970, 42, 115–118 Search PubMed.
- C. M. Hansen, J. Paint Technol., 1967, 39, 505 Search PubMed.
- A. F. M. Barton, Handbook of Solubility Parameters and Other Cohesion Parameters, CRC Press, Inc., Boca Raton, Florida, 1983 Search PubMed.
- A. Martin, P. L. Wu, Z. Liron and D. S. Cohen, J. Pharm. Sci., 1985, 74, 638–642 CAS.
- A. S. R. Anjaneyulu and S. N. Raju, Phytochemistry, 1987, 26, 2805–2810 CrossRef.
- J. Adamcik, V. Castelletto, S. Bolisetty, I. W. Hamley and R. Mezzenga, Angew. Chem., Int. Ed., 2011, 50, 5495–5498 CrossRef CAS.
- J. Schmidtke, W. Stille, H. Finkelmann and S. T. Kim, Adv. Mater., 2002, 14, 746–749 CrossRef CAS.
- J. Schmidtke, S. Kniesel and H. Finkelmann, Macromolecules, 2005, 38, 1357–1363 CrossRef CAS.
- L. Mukhopadhyay, P. K. Bhattacharyya, A. R. Das and S. P. Moulik, Colloid Polym. Sci., 1993, 271, 793–798 CrossRef CAS.
- D. Horn and J. Rieger, Angew. Chem., Int. Ed., 2001, 40, 4330–4361 CrossRef CAS.
- F. Q. Hu, S. P. Jiang, Y. Z. Du, H. Yuan, Y. Q. Ye and S. Zeng, Colloids Surf., B, 2005, 45, 167–173 CrossRef CAS.
- M. E. Matteucci, M. A. Hotze, K. P. Johnston and R. O. Williams, Langmuir, 2006, 22, 8951–8959 CrossRef CAS.
- V. Uskokovic and E. Matijevic, J. Colloid Interface Sci., 2007, 315, 500–511 CrossRef CAS.
- E. T. Pashuck, H. Cui and S. I. Stupp, J. Am. Chem. Soc., 2010, 132, 6041–6046 CrossRef CAS.
- R. Oda, I. Huc, M. Schmutz, S. J. Candau and F. C. MacKintosh, Nature, 1999, 399, 566–569 CrossRef CAS.
- L. Ziserman, H. Y. Lee, S. R. Raghavan, A. Mor and D. Danino, J. Am. Chem. Soc., 2011, 133, 2511–2517 CrossRef CAS.
- J. H. Fuhrhop, P. Schnieder, E. Boekema and W. Helfrich, J. Am. Chem. Soc., 1988, 110, 2861–2867 CrossRef CAS.
- D. S. Chung, G. B. Benedek, F. M. Konikoff and J. M. Donovan, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 11341–11345 CrossRef CAS.
- R. J. H. Hafkamp, B. P. A. Kokke, I. M. Danke, H. P. M. Geurts, A. E. Rowan, M. C. Feiters and R. J. M. Nolte, Chem. Commun., 1997, 545–546 RSC.
- Y. V. Zastavker, N. Asherie, A. Lomakin, J. Pande, J. M. Donovan, J. M. Schnur and G. B. Benedek, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 7883–7887 CrossRef CAS.
- E. Grelet and S. Fraden, Phys. Rev. Lett., 2003, 90, 198302 CrossRef.
- E. Barry, Z. Hensel, Z. Dogic, M. Shribak and R. Oldenbourg, Phys. Rev. Lett., 2006, 96, 018305 CrossRef.
- Z. Dogic and S. Fraden, Curr. Opin. Colloid Interface Sci., 2006, 11, 47–55 CrossRef CAS.
- F. Tombolato, A. Ferrarini and E. Grelet, Phys. Rev. Lett., 2006, 96, 258302 CrossRef.
- K. L. Kohlstedt, F. J. Solis, G. Vernizzi and M. O. de la Cruz, Phys. Rev. Lett., 2007, 99, 030602 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, sample preparation, SAXS, and AFM. See DOI: 10.1039/c1sm06560b |
| This journal is © The Royal Society of Chemistry 2012 |