Flash nanoprecipitation of polystyrene nanoparticles
Chuan
Zhang
,
Vikram J.
Pansare
,
Robert K.
Prud'homme
and
Rodney D.
Priestley
*
Department of Chemical and Biological Engineering, Princeton University, Princeton, New Jersey, USA. E-mail: rpriestl@princeton.edu
First published on 6th October 2011
Abstract
Aside from polymerization techniques, polymer nanoparticles can be generated through the displacement of a solvent with a nonsolvent, i.e., nanoprecipitation. In this study, we utilize a facile process termed Flash NanoPrecipitation (FNP) to generate polystyrene (PS) nanoparticles of several different molecular weights. As compared to PS nanoparticles synthesized by surfactant free emulsion polymerization, nanoparticles prepared by FNP show comparable size distributions when the diameter is less than 150 nm. Furthermore, we illustrate that the sizes of PS nanoparticles prepared by FNP can be fine-tuned by changing the polymer and/or electrolyte concentration. The stabilized nanoparticles contain only the radically polymerized polymer chains, which have sulfate anions at the chain termini and no additional external stabilizers. Calculations of the mechanism of particle formation and stabilization show that the size-dependent electrostatic repulsions between nanoparticles and single collapsed polymer chains control assembly and monodispersity. The ability to independently vary polymer molecular weight and nanoparticle size will enable fundamental studies of the effect of confinement on polymer dynamics in a way not easily achievable by other techniques.
Introduction
Polymeric nanoparticles are vital components of biomedical targeting and diagnostic studies.1–3 Surface functionalization of latex nanoparticles with tightly controlled sizes are used in fluorescent imaging and ligand and antibody targeting.4–6 These applications rely more on the size and surface chemistry of the polymeric nanoparticle than on its core composition. Other applications of polymeric nanoparticles include fillers in thin films for the improvement of thermomechanical properties7 and components in photonic and plasmonic structures.8,9 In these applications, it is the properties of the nanoparticle core that are of paramount importance. Additionally, polymeric nanoparticles can serve as model systems for examining fundamental property changes due to 3-dimensional confinement.10–13 Thus from practical and fundamental viewpoints, the ability to concurrently generate neat polymer nanoparticles (i.e., no residual contaminants), as well as independently controlling nanoparticle size and molecular weight of the polymer core, is highly desirable.Approaches to form polymer nanoparticles can be categorized into three groups: 1) polymerization of monomers (e.g., emulsion, micro-emulsion, mini-emulsion, and interfacial polymerizations),14–17 2) emulsification and solvent stripping to form a “pseudo-latex,”18–20 and 3) nanoprecipitation of a bulk polymer precursor.14,17,21 In typical emulsion polymerization, monomers are combined with an initiator (e.g., ammonium persulfate) and sometimes surfactants (e.g., sodium dodecyl sulfate) in an aqueous medium. The mixture is then allowed to react for several hours to create small emulsions of the polymerizing system. The resulting polymer nanoparticles are monodisperse.15,16 A disadvantage of polymerization techniques is the difficulty of independent control of particle size and molecular weight. That is, both the diameter and the molecular weight increase with increasing extent of polymerization.16 Moreover, residual components, such as monomers, oligomers, and surfactants, can be difficult to remove from these polymerized nanoparticles and may modify the physical properties of the polymer core, thus frustrating measurements of the effect of confinement on polymer nanoparticle properties. In the emulsion stripping process, a polymer solution is emulsified and then the volatile solvent for the polymer is removed to condense and solidify the particle. These techniques, often called “pseudo-latex” formation processes, enable the formation of sub-micron nanoparticles from polymers that cannot be polymerized by normal free-radical polymerization techniques.18–20 However, they suffer from the polydispersity that inherently arises from the size distribution of the parent emulsion drops.
An alternative approach to generate polymer nanoparticles is nanoprecipitation, in which nanoparticles are precipitated from polymer chains in solution as a result of displacing a solvent with a non-solvent.22,23 In the past decade, nanoprecipitation has been primarily used to form biodegradable polymer nanoparticles for drug delivery purposes including poly(lactic acid) (PLA),24,25 poly(lactic-co-glycolic acid) (PLGA),21,26 poly(ε-caprolactone) (PCL),27 and several amphiphilic block copolymers.28–30 One advantage of nanoprecipitation is that since bulk polymer is used as a precursor, independent control of size and molecular weight should be achievable. Additional advantages include fast processing time, low energy consumption, and high reproducibility.31,32 Shortcomings of nanoprecipitation include low mass fraction of nanoparticles in the prepared samples and a broad size distribution for samples with diameters greater than several hundred nanometers.31
Whereas nanoprecipitation has been applied almost exclusively to biodegradable polymers (as evidenced by over 80 publications in the last two years), only a handful of examples on other synthetic polymers exist in the literature.14 Nanoprecipitation of non-biodegradable polymers, such as polystyrene (PS)32,33 and poly(methyl methacrylate) (PMMA),31,32,34 have only started to emerge in the past year. This comes as a surprise since the original patent on nanoprecipitation described the process as a method to generate nanoparticles from a wide-range of polymers and not just biodegradable polymers.22
Here, we utilized a nanoprecipitation process that achieves rapid solvent displacement by means of novel high intensity mixing geometries. The process has been denoted Flash NanoPrecipitation (FNP). The mixing occurs in a central cavity fed by two incoming solvent streams. The geometry and operation of the device has been previously reported35 and has been modeled by computational fluid mechanics.36 Using competitive dye reactions, the mixing time for inlet velocities of ∼1 m/s is on the order of 1.5 ms. In our previous papers, syringe pumps were used to drive the fluid flows at predetermined rates, whereas in this study manual injection was used following the adaptation of Macosko and coworkers.37 Previously, the technique has been used with block copolymer stabilizers to control nanoparticle size, whereas, in this study we use the intrinsic charge of the sulfate-terminated polystyrene chain to produce electrostatic stabilization. As a consequence, sub-150 nm diameter particles of narrow size-distributions can be formed with high reproducibility. In contrast to previous research in the literature which has shown the formation of PS nanoparticles with diameters greater than 200 nm via a more time-consuming dialysis nanoprecipitation technique,32 this work is the first to utilize a simple nanoprecipitation technique to form PS nanoparticles of narrow polydispersity with diameters less than 150 nm. We also illustrate the effect of polymer concentration, polymer molecular weight, and electrolyte (NaCl) concentration on the size and size distribution for nanoparticles prepared by FNP. While in this study we produced only ∼10 mg samples of nanoparticles (an amount sufficient enough to allow for confinement effect studies of the glass transition temperature via differential scanning calorimetry) using manual injection, an advantage of the FNP process is that it is scalable. Using continuous flow in the current geometry would enable production of 3.5 kg/day of material, and currently commercial production of β-carotene nanoparticles are produced at 1400 kg/day using confined impinging jet technology.
Experimental
Material synthesis
Polystyrene nanoparticles were initially synthesized by surfactant-free emulsion polymerization (SFEP), in accordance with procedures described in detail elsewhere.38 Briefly, in a typical synthesis, 0.074 g of ammonium persulfate (Acros Organics, 98%) was initially added to 100 mL of MilliQ H2O in a three-neck flask. The solution was bubbled with N2 for ∼20 min and then heated to 75 °C. Subsequently, 10 g of styrene (Sigma Aldrich, ≥99%) was combined with 0.05 g of acrylic acid (Sigma Aldrich, 99%), and the mixture was injected into the flask. The solution was then allowed to polymerize under reflux conditions for 20 h. The addition of a small amount of acrylic acid as a co-monomer provided additional stability to the colloidal suspension. The molecular weight of the synthesized PS was varied by changing the styrene concentration and/or ammonium persulfate concentration in the reactor. After synthesis, the PS nanoparticles were washed twice with MilliQ H2O through centrifugation and ultimately suspended in water. A portion of the water-suspended sample was subsequently dried and annealed at 150 °C for 12 h to form bulk polymer, which was then used as the precursor polymer for Flash NanoPrecipitation.Fig. 1a shows a schematic of the FNP process employed to precipitate PS nanoparticles, while Fig. 1b shows the actual FNP setup employed in the laboratory. A confined impinging jet mixer composed of two separate streams was used. Here, a syringe containing 3 mL of the synthesized bulk PS dissolved in tetrahydrofuran (THF) was placed at the inlet of Stream 1, and a syringe containing 3 mL of pure MilliQ H2O or a H2O/NaCl solution was placed at the inlet of Stream 2. Subsequently, fluid was expressed manually from both syringes at the same rate (∼3 ml in ∼3 s), causing the two streams to merge into a mixing stream. The flow rates through the mixer were kept approximately constant at 2 mL/s, which resulted in a jet velocity of ∼1 m/s through the 0.5 mm diameter orifices and a Reynolds number35 of ∼3500. The mixed exit stream was then diluted into a 27 mL water reservoir, which quenched the precipitated nanoparticles. The precipitated PS nanoparticles suspended in an aqueous solution were collected in scintillation vials. THF was subsequently removed through membrane dialysis or air evaporation over the course of 48 h.
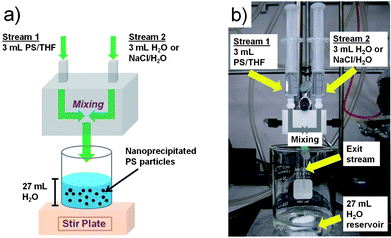 | ||
| Fig. 1 (a) Schematic and (b) actual laboratory setup of the FNP mixing process to generate PS nanoparticles. | ||
Characterization
Weight-average molecular weight (Mw) and number-average molecular weight (Mn) of each polymer sample created from SFEP was determined using gel permeation chromatography (Waters 515 HPLC pump, Eppendorf CH-460 column heaters, Waters 410 differential refractometer, Agilent PLgel mixed B column, THF eluent). Z-average diameters, size distribution curves, and polydispersity indices (PDI) of size distributions of PS nanoparticles suspended in water from SFEP and FNP were determined using dynamic light scattering (DLS) (Malvern Instruments Zetasizer Nano-ZS ZEN 3600). Zeta potentials of nanoparticle samples were obtained using the same instrument (Malvern Instruments Zetasizer Nano-ZS ZEN 3600) in a disposable folded capillary cell. The instrument performs laser Doppler velocimetry to obtain the electrophoretic mobility, which is then converted to a zeta potential via the Henry equation. Images of PS nanoparticles were obtained using scanning electron microscopy (SEM) (FEI XL-30). Prior to imaging, nanoparticles were drop-casted onto carbon tape, allowed to dry in the hood for 12 h, and then coated with a 5 nm thick iridium layer.Results and discussion
Experimental
The basic premise behind Flash NanoPrecipitation is that rapid micromixing produces solvent exchange between good solvent and anti-solvent conditions in a time scale faster than the aggregation time of the solute. The process is shown schematically in Fig. 1. The solvent stream containing PS dissolved in THF (Stream 1) was rapidly mixed with an incoming water stream (Stream 2), which caused collapse of the hydrophobic polymer chain in the aqueous solvent and subsequent aggregation. The surfaces of the nanoparticles were negatively charged, as determined from zeta-potential measurements (ζ ∼ −40–50 mV). The inherent negative surface charge, resulting from sulfate-terminated polymer chains from the ammonium persulfate initiator in emulsion polymerization, provided stability to the nanoprecipitated PS particles, which will be discussed below.The effect of PS concentration in THF on the size of polymer nanoparticles formed via FNP was determined by varying the concentration between 0.5 mg/mL to 10 mg/mL in Stream 1 of the mixer for PS of Mw = 376 kg/mol. Fig. 2a-c show representative SEM images of the nanoprecipitated PS particles using concentrations of 2 mg/mL, 5 mg/mL and 10 mg/mL, respectively. It was observed that as the concentration increased, the particle size increased. Additionally, the precipitated particles were fairly monodisperse up until a concentration of 5 mg/mL. At a concentration of 10 mg/mL (which corresponded to a size of ∼140 nm), the precipitated particles became more polydisperse and exhibited a broader size distribution.
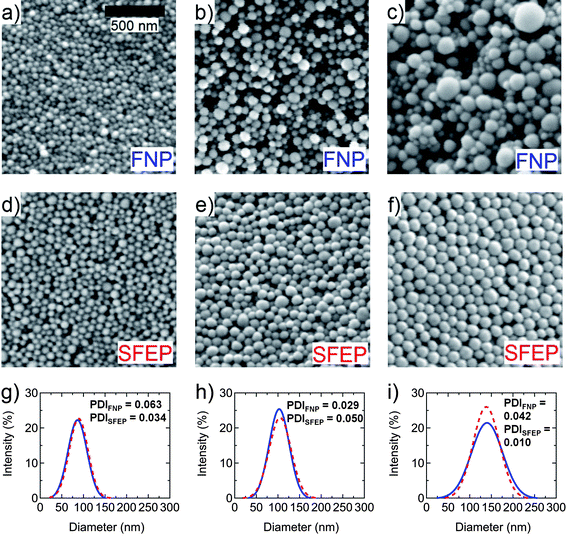 | ||
| Fig. 2 Representative SEM images of PS nanoparticles (Mw = 376 kg/mol) made from PS concentrations of (a) 2 mg/mL, (b) 5 mg/mL, and (c) 10 mg/mL in Stream 1 of the FNP mixing process, with Stream 2 containing pure H2O. SEM images (d)-(f) show similar-sized PS nanoparticles synthesized from SFEP, in comparison to (a)–(c), respectively. Solid lines in (g)–(i) show size distribution curves of PS nanoparticles from FNP in images (a)–(c), respectively, whereas dashed lines in (g)–(i) represent size distribution curves of PS nanoparticles from SFEP in images (d)–(f), respectively. | ||
Fig. 2d–f show SEM images of PS nanoparticles from SFEP, roughly corresponding to sizes of particles in Fig. 2a–c, respectively. By comparing Fig. 2a to 2d, 2b to 2e, and 2c to 2f, it was observed that for particles with diameters less than 140 nm, i.e., for PS concentrations of 5 mg/mL or less in FNP, the FNP technique generated nanoparticles as monodisperse as particles prepared by SFEP. For larger particles, i.e., for PS concentration of 10 mg/mL in FNP, the sizes of particles from FNP became polydisperse, whereas particles from SFEP remained monodisperse. Thus, one of the major challenges for nanoprecipitation techniques is maintaining low polydispersity for larger particles. A quantitative comparison of size polydispersity between nanoparticles from FNP and SFEP was made by plotting size distribution curves (determined from DLS), as shown in Fig. 2g–i. The solid lines correspond to nanoparticles from FNP, while dashed lines correspond to nanoparticles from SFEP. These size distribution curves, along with corresponding PDI values, provide further evidence that monodispersity, i.e., a narrow size distribution, can be maintained for PS nanoparticles from FNP with diameters less than 140 nm.
To examine the effect of polymer molecular weight on nanoparticle size, two additional PS molecular weights (Mw = 92 kg/mol and Mw = 770 kg/mol) were used in the FNP technique. Note that the molecular weight polydispersity indices (i.e., Mw/Mn) for the three samples were all ∼3.5, as determined from GPC. Fig. 3a plots the z-average diameter (determined from DLS) of PS nanoparticles as a function of PS concentration in Stream 1 of the FNP mixing process for the three different molecular weights. For each molecular weight, the diameter of PS nanoparticles increased linearly with increasing PS concentration in Stream 1, in agreement with the SEM images in Fig. 2. At the same polymer concentration, the particle size increased with increasing molecular weight. This increase in size with molecular weight may be explained by the increased size of the collapsed polymer chain and lower charge density contributed by each chain, resulting in larger particles when these aggregate. From Fig. 3a, it becomes clear that PS nanoparticle size and molecular weight can be controlled independently in a straightforward manner. For example, ∼90 nm diameter nanoparticles with molecular weights of 92 kg/mol, 376 kg/mol, and 770 kg/mol can be made using PS concentrations of 10 mg/mL, 5 mg/mL, and 0.5 mg/mL, respectively.
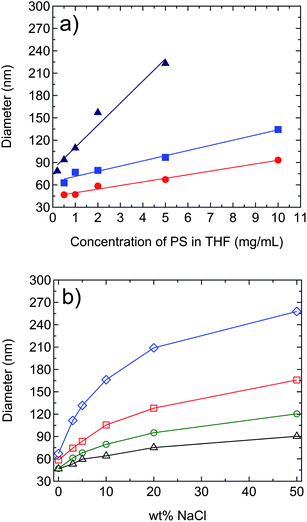 | ||
Fig. 3 (a) Change in PS nanoparticle diameter as a function of PS concentration in Stream 1 of the FNP mixing process for three different PS molecular weights: 92 kg/mol ( ), 376 kg/mol ( ), 376 kg/mol ( ), and 770 kg/mol ( ), and 770 kg/mol ( ), with pure H2O in Stream 2. (b) Change in PS nanoparticle diameter as a function of wt% of NaCl in Stream 2 of the FNP mixing process for different PS (Mw = 92 kg/mol) concentrations in Stream 1: 0.5 mg/mL (△), 1 mg/mL ( ), with pure H2O in Stream 2. (b) Change in PS nanoparticle diameter as a function of wt% of NaCl in Stream 2 of the FNP mixing process for different PS (Mw = 92 kg/mol) concentrations in Stream 1: 0.5 mg/mL (△), 1 mg/mL ( ), 2 mg/mL ( ), 2 mg/mL ( ), and 5 mg/mL ( ), and 5 mg/mL ( ). Size measurement errors are within the marker size. Solid lines are guides for the eye. ). Size measurement errors are within the marker size. Solid lines are guides for the eye. | ||
An additional variable in tuning the size of polymer nanoparticles prepared from FNP was explored, i.e., adding NaCl to Stream 2 of the mixer. Fig. 3b shows the change in diameter of PS nanoparticles (Mw = 92 kg/mol) for different PS concentrations in Stream 1 as the wt% of NaCl (with respect to PS weight) was varied in Stream 2 from 0 wt% to 50 wt%. As the wt% of NaCl increased, the diameter of the particles increased at each PS concentration. Hence, by changing PS concentration and/or wt% of NaCl, the diameter of PS nanoparticles prepared from FNP can be precisely controlled. Note that similar trends were observed for the other PS molecular weights, i.e., for Mw = 376 kg/mol and Mw = 770 kg/mol. Clearly, at certain unique values of PS concentration and/or wt% of NaCl for each molecular weight, similar-sized nanoparticles with different molecular weights can be generated from the FNP technique. We note that for similar-sized nanoparticle prepared with and without NaCl, the width of the size distribution curves were similar.
Fig. 4 shows representative SEM images of PS nanoparticles (Mw = 92 kg/mol) made using 0 wt%, 3 wt%, 5 wt%, 10 wt%, 20 wt%, and 50 wt% of NaCl (with respect to PS weight) in Stream 2 and 2 mg/mL PS concentration in Stream 1 of the FNP mixing process. From these images, it is clear that diameters increased with increasing wt% of NaCl, in agreement with the trend observed in Fig. 3b. For the particular case, i.e., Mw = 92 kg/mol and 2 mg/mL PS concentration, PS nanoparticles appeared to be fairly monodisperse, up until 50 wt% of NaCl (with respect to PS weight).
 | ||
| Fig. 4 Representative SEM images of PS nanoparticles (Mw = 92 kg/mol) made using (a) 0%, (b) 3%, (c) 5%, (d) 10%, (e) 20%, and (f) 50% wt% of NaCl (with respect to PS weight) in Stream 2 and 2 mg/mL PS concentration in Stream 1 of the FNP mixing process. | ||
Mechanism of assembly
The mechanisms involved in the Flash NanoPrecipitation of high molecular weight polymer are interestingly and subtly different than the mechanisms of controlled assembly of small hydrophobic organic molecules with stabilizing block copolymers, i.e., the normal mode of operation of FNP. The differences are twofold. First, in small molecule nanoparticle formation, the solvent exchange causes supersaturation, but there is an activation barrier to assembly, i.e., a nucleation step.39 Particles must first grow larger than the critical nuclei size and then the process becomes growth controlled. In systems that are nucleation controlled, increasing nucleation rate decreases the final nanoparticle size because the solute deposits on the larger number of nuclei that are formed. In contrast, if the system is growth controlled, then a higher concentration of solute means that particles grow larger the longer they are in the growth regime, before the stabilizing block copolymer quenches growth.31,40 For a 92 kg/mol polymer chain, the collapsed radius is 3.3 nm, as calculated from the mass and density of a single PS chain. This is much larger than a critical nuclei size; therefore, there is no barrier to nucleation. The nanoparticle size is controlled only by micro-mixing to ensure homogeneous polymer distribution in space and by growth kinetics.The second difference is in the mechanism of stabilization. For traditional FNP, the adsorption of the block copolymer on the nanoparticle surface determines the end of aggregation and freezes the size. In the current process, there is no external stabilizing agent. Each polymer chain, which has been initiated by a sulfate radical, carries with it a unit negative charge. The stronger electrostatic repulsion between two nanoparticles relative to the weaker repulsion between a nanoparticle and a collapsed polymer chain (unimer) controls growth and stabilization of nanoparticles. The following illustrative calculations demonstrate the essential features of the process and highlight the reason why SFEP produces narrower particle size distributions at larger nanoparticle sizes than does FNP. The measured electrophoretic mobilities of PS nanoparticles produced by FNP from 92 kg/mol PS in varying ionic strength solutions are shown in Fig. 5. Precipitations were conducted at 1 mg/mL PS concentration in the THF stream. As shown, nanoparticle size increased with ionic strength up to 1 × 10−1 M at which point the nanoparticle dispersion flocculated upon formation, and stable particles could no longer be produced. The zeta potential of the nanoparticles was measured to be −39 mV at 10−5 M and becomes more negative (−55 mV) at 5 × 10−2 M. Subsequently, the surface charge diminishes (i.e., trends toward zero) at higher ionic strengths. The increase and then decrease in mobility with ionic strength has been previously reported for similar surfactant free, charge stabilized latexes.41,42 This is in contrast to a sharp monotonic decrease in surface potential with ionic strength that would be expected if the PS surface had a fixed density of sulfate groups that determined the surface potential.43 The origin of the ionic-strength-dependent surface charge is an open debate, but reports of negative potentials on hydrophobic surfaces generally support a mechanism involving the more weakly solvated anions interacting with the predominantly hydrophobic surface and giving a zeta potential that reflects this anionic charging.42,44 The surface charge decreases at even higher ionic strength due to conventional electrostatic screening.42,43
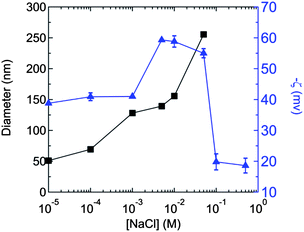 | ||
Fig. 5 Dependence of zeta potential (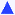 ) and size (■) on NaCl concentration for PS (Mw = 92 kg/mol) nanoparticles generated at a polymer concentration of 1 mg/mL in Stream 1 of the FNP process. The 0.1 M and 0.5 M samples aggregated upon formation, thus their sizes are not shown. The (negative) zeta potential passes through a maximum and decreases (tends towards zero) at higher ionic strength. ) and size (■) on NaCl concentration for PS (Mw = 92 kg/mol) nanoparticles generated at a polymer concentration of 1 mg/mL in Stream 1 of the FNP process. The 0.1 M and 0.5 M samples aggregated upon formation, thus their sizes are not shown. The (negative) zeta potential passes through a maximum and decreases (tends towards zero) at higher ionic strength. | ||
The surface charge density (σ) can be calculated from the Graham equation:43
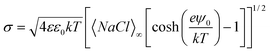 | (1) |
The interaction energy (WTotal) between two 200 nm diameter PS nanoparticles having the experimentally determined surface potentials (Fig. 5) and the energy between a 200 nm nanoparticle and a unimer chain are calculated from the balance of electrostatic repulsions (WElectrostatic) and attractive London van der Waals interactions (WVDW):
| WTotal = WElectrostatic + WVDW | (2) |
The electrostatic interactions between two spheres of radii R1 and R2 (m) with different surface potentials has been derived by Hogg et al.45 and Israelachivili:43
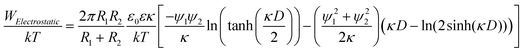 | (3) |
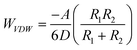 | (4) |
The total interaction energies, WTotal, are shown in Fig. 6. From Fig. 6a, at low ionic strength (10−5 M), the much weaker electrostatic repulsions between the unimer polymer chain and nanoparticle cause the potential energy barrier to be only 4 kT, whereas the barrier for nanoparticle-nanoparticle interaction is 105 kT. In 10−3 M NaCl, the nanoparticle-nanoparticle barrier is still 99 kT, but the unimer-nanoparticle interaction is entirely attractive (Fig. 6b). For 10−1 M NaCl, the nanoparticle-nanoparticle interactions become attractive (Fig. 6c). The aggregation of single collapsed polymer chains onto either larger nanoparticles or with themselves proceeds at all ionic strengths, but the aggregation of larger nanoparticles amongst themselves is prevented at lower ionic strengths. Therefore, nanoparticles produced at ionic strengths below 10−1 M would be expected to be stable, while nanoparticles produced at or above 10−1 M ionic strengths would be expected to be unstable. This is what is observed experimentally, as shown in Fig. 5. The dependence of nanoparticle size on polymer concentration in the FNP process (as shown in Fig. 3a) can also be explained by considering the interaction potential at low ionic strength in Fig. 6a. The potential barrier is low enough that unimers or small aggregates can continue to add to nanoparticles that are larger than the critical size for electrostatic stability. The growth occurs by continued addition when there is a higher concentration of unimers. The effect of polymer molecular weight on nanoparticle size could be addressed by the model by conducting the calculations with “unimers” with sizes that scale with molecular weight. To demonstrate the physics behind the process, we have selected the unimer size based only on the 92 kg/mol polymer.
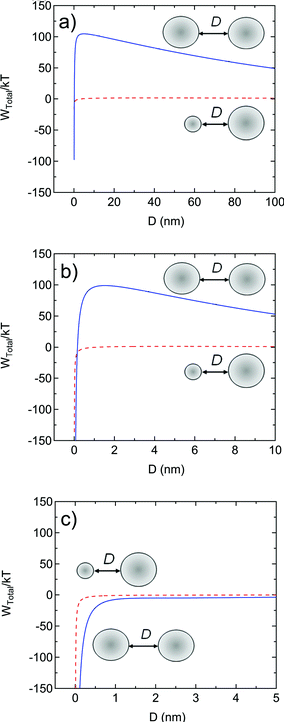 | ||
| Fig. 6 Total interaction potential, Wtotal/kT, as a function of distance between two 200 nm diameter PS spheres (solid blue lines) and between a 200 nm sphere and a 3.3 nm unimer (dashed red lines) as calculated from eqn (2)–(4) at ionic strengths of (a) 10−5 M (0.64 wt% of NaCl with respect to PS) with the experimentally measured zeta potential of −39 mV for the 200 nm sphere and −15 mV for the unimer sphere, (b) 10−3 M (64 wt% of NaCl with respect to PS) with the experimentally measured zeta potential of −41 mV for the 200 nm sphere and −16 mV for the unimer sphere, and (c) 10−1 M (6400 wt% of NaCl with respect to PS) with the experimentally measured zeta potential of −20 mV for the 200 nm sphere and −8 mV for the unimer sphere. At all ionic strengths, the repulsive barrier is never high enough to prevent unimer from aggregating with the 200 nm spheres, but the repulsive barrier is large enough at 10−5 M and 10−3 M to prevent 200 nm sphere from aggregating, thus ensuring colloidal stability. At 10−1 M, the potential interaction between 200 nm spheres is entirely attractive and aggregation would be expected as is seen experimentally (Fig. 5). | ||
The increase in polydispersity for nanoparticles with increasing size can also be understood in terms of the process of aggregation. In our previous study of the FNP assembly of 3 nm hydrophobic Au colloids, i.e., the same size as the collapsed polymer chain unimers considered here, the size and polydispersity were quantitatively modeled using diffusion limited aggregation.46 The cutoff time in the previous study was the time scale for polymer stabilization. This mechanism is consistent with the results seen for the current polymer nanoparticles. The lack of sufficient electrostatic stabilization enables particles below a critical electrostatic stabilization size to aggregate. The random aggregation process creates a polydisperse distribution of sizes. However two nanoparticles slightly below the critical size for stability can aggregate to form a stable nanoparticle that is significantly larger than the minimum critical size. The model of random aggregation46 predicts that the polydispersity of the distribution increases with the number of random aggregation steps; that is, the polydispersity increases with increasing size. In contrast, SFEP involves a growth process where an initial number of nuclei are formed and then growth occurs by single chain addition; the rate of which is equal for every nuclei in the population. This single chain addition results in a population that is self sharpening and becomes more monodisperse with increasing size until other destabilizing mechanisms come into play.47, 48 Modeling the concentration dependence of nanoparticle size would involve a population balance approach that would capture both the dynamics of assembly and the electrostatic energy barrier that varies with nanoparticle size in the population.49–51
The mechanism of electrostatic control of nanoparticle size during FNP assembly is further demonstrated in Fig. 7. Nanoparticles were initially generated from 376 kg/mol PS using 10 mg/ml PS concentration in Stream 1 of the FNP process and 3 wt% of NaCl (with respect to PS weight) in Stream 2 to give a final ionic strength of 4.7 × 10−4 M in the nanoparticle solution. This original sample was subsequently diluted into NaCl solutions to prepare a set of samples over the ionic strength range of ∼ 10−5 - 2 M. The larger size for the nanoparticles in Fig. 7 relative to Fig. 5 arises because of the higher molecular weight of the PS, as well as a higher concentration of PS in Stream 1 of the FNP process, used in this formulation. Since nanoparticles grow in size until electrostatic repulsions create interaction potential barriers high enough to prevent further aggregation, the nanoparticles are at a critical size in a medium of that ionic strength. When the nanoparticles are placed in a medium of lower ionic strength, the electrostatic repulsions are increased and the nanoparticles are stable and their size is unaffected. However, when the ionic strength is increased from the synthesis conditions of 4.7 × 10−4 M to 0.51 M, electrostatic repulsions are decreased and the nanoparticles aggregate to form clusters of 600 nm. At 1.7 M, the nanoparticles aggregate to 1000 nm. This process of subsequently destabilizing a nanoparticle dispersion that has been stable under low ionic strength is fundamentally different than the rapid kinetic assembly of nanoparticles under conditions where particle stability is never achieved (Fig. 5, 10−1 M and above). What is unexpected is that the aggregates that are produced at these higher ionic strengths are not gross precipitates but have a well defined aggregation size. This phenomenon may be related to the recent discovery of large clusters that can be formed by balancing short ranged attractive forces with longer range cooperative electrostatic forces.52–54
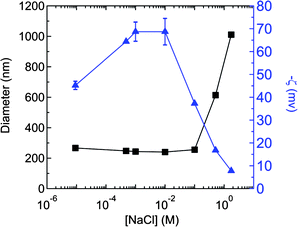 | ||
Fig. 7 Dependence of zeta potential ( ) and size (■) on NaCl concentration for PS (Mw = 376 kg/mol) nanoparticles generated at 4.7 × 10−4 M (3 wt% with respect to PS) NaCl using a polymer concentration of 10 mg/mL in Stream 1 of the FNP process. The original sample formed at 4.7 × 10−4 M NaCl was subsequently diluted into NaCl solutions to prepare a set of samples over the ionic strength range of ∼10−5 - 2 M. Particle size was unaffected by ionic strength below 10−1 M, but aggregation was observed above this ionic strength. The (negative) zeta potential increases and then decreases over this range. ) and size (■) on NaCl concentration for PS (Mw = 376 kg/mol) nanoparticles generated at 4.7 × 10−4 M (3 wt% with respect to PS) NaCl using a polymer concentration of 10 mg/mL in Stream 1 of the FNP process. The original sample formed at 4.7 × 10−4 M NaCl was subsequently diluted into NaCl solutions to prepare a set of samples over the ionic strength range of ∼10−5 - 2 M. Particle size was unaffected by ionic strength below 10−1 M, but aggregation was observed above this ionic strength. The (negative) zeta potential increases and then decreases over this range. | ||
Understanding the mechanism of nanoparticles growth by FNP versus SFEP suggests future research directions to form larger FNP nanoparticles with narrower size distributions. Since unimer addition to monodisperse seeds produces narrow distributions, it may be possible to use multi-stage FNP mixers to sequentially add unimers to narrowly distribute seed nanoparticles with initial sizes below 150 nm. It should also be possible to produce layered structures with polymers of different compositions or molecular weights using multi-stage mixers. In one sense we have already demonstrated this capability by coating SFEP nanoparticles with an amphiphilic block copolymer.55
Conclusion
In summary, we utilized a facile method termed Flash NanoPrecipitation to successfully generate PS nanoparticles of wide-ranging sizes and different molecular weights. As compared to PS nanoparticles synthesized from surfactant free emulsion polymerization, nanoparticles prepared from FNP showed comparable size-distributions when the diameters of the particles were under 150 nm. Furthermore, we showed that the nanoparticle size can be fine-tuned by changing the PS concentration and/or changing wt% of NaCl in the FNP technique, i.e., an increase in PS concentration and/or wt% of NaCl led to an increase in particle size. The mechanism of nanoparticles growth and stability can be understood by considering the competition between attractive London Van der Waals attraction and electrostatic repulsion. The ability to rapidly produce a range of nanoparticle sizes from a range of polymer precursors with no added stabilizing components or residual impurities will greatly facilitate the studies of polymer dynamics in a 3-dimensionally confined geometry.Acknowledgements
We acknowledge usage of the PRISM Imaging and Analysis Center, which is supported in part by the NSF MRSEC program through the Princeton Center for Complex Materials (DMR-0819860). R.D.P. acknowledges the donors of the American Chemical Society Petroleum Research Fund (PRF 49903-DNI10) and the 3M-nontenured faculty grant program for partial support of this work.References
- K. Kempe, A. Vollrath, H. W. Schaefer, T. G. Poehlmann, C. Biskup, R. Hoogenboom, S. Hornig and U. S. Schubert, Macromol. Rapid Commun., 2010, 31, 1869–1873 CrossRef CAS
.
- G. Gaucher, R. H. Marchessault and J. C. Leroux, J. Controlled Release, 2010, 143, 2–12 CrossRef CAS
- C. E. Mora-Huertas, H. Fessi and A. Elaissari, Int. J. Pharm., 2010, 385, 113–142 CrossRef CAS
- M. E. Gindy and R. K. Prud'homme, Expert Opin. Drug Delivery, 2009, 6, 865–878 CrossRef CAS
- A. Vollrath, S. Schubert, N. Windhab, C. Biskup and U. S. Schubert, Macromol. Rapid Commun., 2010, 31, 2053–2058 CrossRef CAS
- N. Ozturk, N. Bereli, S. Akgol and A. Denizli, Colloids Surf., B, 2008, 67, 14–19 CrossRef CAS
- S. M. Liff, N. Kumar and G. H. McKinley, Nat. Mater., 2006, 6, 76–83 CrossRef
- T. Still, W. Cheng, M. Retsch, U. Jonas and G. Fytas, J. Phys.: Condens. Matter, 2008, 20, 404203 CrossRef
- E. Ozbay, Science, 2006, 311, 189–193 CrossRef CAS
- T. Sasaki, A. Shimizu, T. H. Mourey, C. T. Thurau and M. D. Ediger, J. Chem. Phys., 2003, 119, 8730–8735 CrossRef CAS
- Y. Rharbi, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2008, 77, 031806 CrossRef
- C. Zhang, Y. Guo and R. D. Priestley, Macromolecules, 2011, 44, 4001–4006 CrossRef CAS
- Y. Guo, C. Zhang, C. Lai, R. D. Priestley, M. D'Acunzi and G. Fytas, ACS Nano, 2011, 5, 5365–5373 CrossRef CAS
- S. Schubert, J. T. Delaney and U. S. Schubert, Soft Matter, 2011, 7, 1581–1588 RSC
-
G. G. Odian, Principles of Polymerization, Wiley-Interscience, Hoboken, N.J., 2004 Search PubMed
- K. Tauer, R. Deckwer, I. Kuhn and C. Schellenberg, Colloid Polym. Sci., 1999, 277, 607–626 CAS
- J. P. Rao and K. E. Geckeler, Prog. Polym. Sci., 2011, 36, 887–913 CrossRef CAS
- O. Thioune, H. Fessi, J. P. Devissaguet and F. Puisieux, Int. J. Pharm., 1997, 146, 233–238 CrossRef CAS
- D. Quintanar-Guerrero, E. Allemann, H. Fessi and E. Doelker, Int. J. Pharm., 1999, 188, 155–164 CrossRef CAS
- N. Anton, J. P. Benoit and P. Saulnier, J. Controlled Release, 2008, 128, 185–199 CrossRef CAS
- T. Govender, S. Stolnik, M. C. Garnett, L. Illum and S. S. Davis, J. Controlled Release, 1999, 57, 171–185 CrossRef CAS
-
H. Fessi, J. P. Devissaguet, F. Puisieux and C. Thies, US Pat., 1990, 5118528 Search PubMed
- H. Fessi, F. Puisieux, J. P. Devissaguet, N. Ammoury and S. Benita, Int. J. Pharm., 1989, 55, R1–R4 CrossRef CAS
- E. Leo, B. Brina, F. Forni and M. A. Vandelli, Int. J. Pharm., 2004, 278, 133–141 CrossRef CAS
- P. Legrand, S. Lesieur, A. Bochot, R. Gref, W. Raatjes, G. Barratt and C. Vauthier, Int. J. Pharm., 2007, 344, 33–43 CrossRef CAS
- M. S. Muthu, M. K. Rawat, A. Mishra and S. Singh, Nanomed.: Nanotechnol., Biol. Med., 2009, 5, 323–333 CrossRef CAS
- F. Lince, D. L. Marchisio and A. A. Barresi, J. Colloid Interface Sci., 2008, 322, 505–515 CrossRef CAS
- B. K. Johnson and R. K. Prud'homme, Phys. Rev. Lett., 2003, 91, 118302 CrossRef
- J. Ren, H. Y. Hong, J. X. Song and T. B. Ren, J. Appl. Polym. Sci., 2005, 98, 1884–1890 CrossRef CAS
- F. Quaglia, L. Ostacolo, G. De Rosa, M. I. La Rotonda, M. Ammendola, G. Nese, G. Maglio, R. Palumbo and C. Vauthier, Int. J. Pharm., 2006, 324, 56–66 CrossRef CAS
- J. Aubry, F. Ganachaud, J. P. C. Addad and B. Cabane, Langmuir, 2009, 25, 1970–1979 CrossRef CAS
- S. Hornig, T. Heinze, C. R. Becer and U. S. Schubert, J. Mater. Chem., 2009, 19, 3838–3840 RSC
- N. Miletic, V. Abetz, K. Ebert and K. Loos, Macromol. Rapid Commun., 2010, 31, 71–74 CrossRef CAS
- I. Perevyazko, A. Vollrath, S. Hornig, G. M. Pavlov and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3924–3931 CrossRef CAS
- B. K. Johnson and R. K. Prud'homme, AIChE J., 2003, 49, 2264–2282 CrossRef CAS
- Y. Liu and R. O. Fox, AIChE J., 2006, 52, 731–744 CrossRef CAS
-
C. W. Macosko, University of Minnesota, personal communication, 2011
- L. Zhang, M. D'Acunzi, M. Kappl, G. K. Auernhammer, D. Vollmer, C. M. van Kats and A. van Blaaderen, Langmuir, 2009, 25, 2711–2717 CrossRef CAS
- D. Horn and J. Rieger, Angew. Chem., Int. Ed., 2001, 40, 4330–4361 CrossRef CAS
- Y. Liu, K. Kathan, W. Saad and R. K. Prud'homme, Phys. Rev. Lett., 2007, 98, 036102 CrossRef
- M. Elimelech and C. R. Omelia, Colloids Surf., 1990, 44, 165–178 CrossRef CAS
- C. F. Zukoski and D. A. Saville, J. Colloid Interface Sci., 1985, 107, 322–333 CrossRef CAS
-
J. N. Israelachvili, Intermolecular and Surface Forces, Academic Press, Burlington, MA, 2011 Search PubMed
- B. R. Midmore and R. J. Hunter, J. Colloid Interface Sci., 1988, 122, 521–529 CrossRef CAS
- R. Hogg, T. W. Healy and D. W. Fuersten, Trans. Faraday Soc., 1966, 62, 1638–1651 RSC
- M. E. Gindy, A. Z. Panagiotopoulos and R. K. Prud'homme, Langmuir, 2008, 24, 83–90 CrossRef CAS
- S. C. Thickett and R. G. Gilbert, Polymer, 2007, 48, 6965–6991 CrossRef CAS
- The details of the “single chain event” depend on the exact locus of polymerization. If the polymerization occurs in the aqueous phase, then the polymer collapses and diffuses until it aggregates with an existing nanoparticle. If the locus is inside the nanoparticle with monomer solubilized into the core of the nanoparticle, then polymerization initiates with the entry of a single radical into the nanoparticle core and continues until the next radical enters; the radicals are terminated by recombination. Thus, the polymer mass addition is by a “single chain“ or unimer depending on the mechanism. See Ref. 46 for a thorough review of emulsion polymerization mechanisms.
- J. C. Cheng and R. O. Fox, Ind. Eng. Chem. Res., 2010, 49, 10651–10662 CrossRef CAS
- J. C. Cheng, R. D. Vigil and R. O. Fox, J. Colloid Interface Sci., 2010, 351, 330–342 CrossRef CAS
- E. Gavi, D. L. Marchisio, A. A. Barresi, M. G. Olsen and R. O. Fox, Chem. Eng. Res. Des., 2010, 88, 1182–1193 CrossRef CAS
- L. Porcar, P. Falus, W. R. Chen, A. Faraone, E. Fratini, K. L. Hong, P. Baglioni and Y. Liu, J. Phys. Chem. Lett., 2010, 1, 126–129 CrossRef CAS
- M. M. van Schooneveld, V. W. de Villeneuve, R. P. Dullens, D. G. Aarts, M. E. Leunissen and W. K. Kegel, J. Phys. Chem. B, 2009, 113, 4560–4564 CrossRef CAS
-
K. P. Johnston, University of Texas – Austin, personal communication, 2011
- S. J. Budijono, B. Russ, W. Saad, D. H. Adamson and R. K. Prud'homme, Colloids Surf., A, 2010, 360, 105–110 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2012 |