Enantio- and diastereoselective addition of cyclohexyl Meldrum's acid to β- and α,β-disubstituted nitroalkenesvia N-sulfinyl urea catalysis†
Kyle L.
Kimmel
,
Jimmie D.
Weaver
and
Jonathan A.
Ellman
*
Department of Chemistry, Yale University, Connecticut 06520, USA. E-mail: jonathan.ellman@yale.edu; Tel: 203-432-2647
First published on 25th August 2011
Abstract
Using N-sulfinyl urea catalysis, a method has been developed for the asymmetric synthesis of biologically important γ-amino acids with a high level of efficiency, practicality and unprecedented control of multiple stereocenters. This method is based upon the highly enantio- and diastereoselective addition of cyclohexyl Meldrum's acid as an easily deprotectable monocarboxylic acid equivalent. The addition to both β-substituted and α,β-disubstituted nitroalkenes using N-sulfinyl urea organocatalyst 8 is described. The utility of this new method toward drug production is demonstrated by the mole scale preparation of a key precursor to the commercial drug Lyrica using catalyst 8 at only 0.2 mol% loading. Moreover, α,β-disubstituted nitroalkene addition products were efficiently converted to γ-amino acid derivatives without epimerization of either stereocenter.
Introduction
γ-Amino acids are present in numerous drugs, drug candidates and bioactive natural products (Fig. 1).1,2 Moreover, this structure can readily be transformed into pyrrolidinones and pyrrolidines, which are also present in a large number of bioactive compounds.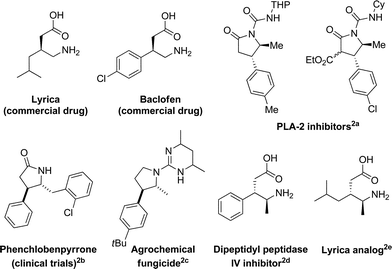 | ||
| Fig. 1 Representative examples of bioactive γ-amino acid derivatives. | ||
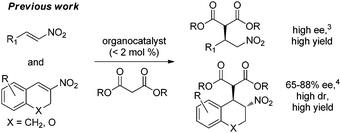
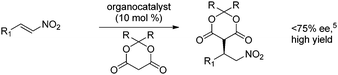
Consequently, highly effective organocatalytic methods have been developed for the asymmetric addition of malonates to β-substituted nitroalkenes at low catalyst loading to provide efficient and practical access to γ-amino acid derivatives (eqn 1).3 In contrast, for additions to α,β-disubstituted nitroalkenes only a narrow set of cyclic substrates have been reported.4 Moreover, despite considerable effort, only moderate enantioselectivity has been achieved for the organocatalytic addition of Meldrum's acid derivatives to nitroalkenes (eqn 2).5 For each of the reported examples, high catalyst loading was also used.
 | (1) |
 | (2) |
 | (3) |
The successful asymmetric addition of Meldrum's acid with high selectivity and catalyst efficiency is a worthwhile and important goal due to two distinctive properties of the Meldrum's acid structure. First, Meldrum's acid derivatives are more acid labile than malonates due to their acetal framework, thereby enabling a mild and convenient single step, acid-catalyzed process for tandem deprotection/decarboxylation to form the corresponding γ-amino acid derivatives.6 Second, Meldrum's acid derivatives (pKa in DMSO = 7–8)7 are considerably more acidic than malonates (pKa in DMSO = 16–17),7 resulting in new opportunities for reactivity and selectivity. In particular, the previously reported addition of malonates to cyclic nitroalkenes (eqn 1), presumably provides the more stable trans-disubstituted products through thermodynamic control as a consequence of the comparable acidity of the malonate nucleophile and the nitroalkane product (pKa in DMSO = 16–17).7 Unfortunately, only a very modest thermodynamic ratio of trans-/cis-isomers for acyclic nitroalkene addition products with α- and β-stereocenters is likely to be observed (see below). The much greater acidity of Meldrum's acids relative to both malonates and nitroalkanes should enable efficient kinetic control and potentially high selectivity for additions to α,β-disubstituted nitroalkenes.8
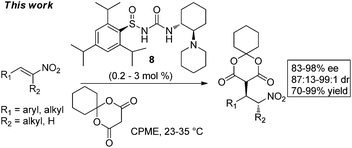
Herein, we report that N-sulfinyl urea9,10catalyst 8 at low catalyst loadings (0.2–3 mol%) provides the first highly enantio- and diastereoselective organocatalytic addition of Meldrum's acid derivatives to nitroalkenes (eqn 3). This work also provides the first example of the organocatalytic addition of any carbon nucleophile to acyclic α,β-disubstituted nitroalkenes to set both the α- and β-stereocenters in acyclic products with high enantio- and diastereoselectivity.11 Significantly, the Meldrum's acid addition products undergo facile hydrolysis and decarboxylation under acidic conditions followed by reduction to directly afford enantioenriched γ-amino acids without epimerization. A representative addition product of Meldrum's acid to an α,β-disubstituted nitroalkene has also been converted to useful γ-amino acid-derivatives without epimerization. The practicality of our method is further demonstrated by the mole scale addition of cyclohexyl Meldrum's acid to nitroalkene 1b at only 0.2 mol% organocatalyst loading followed by one-step hydrolysis/decarboxylation to provide a known precursor to the drug S-Pregabalin (Lyrica),12 which is used extensively for the treatment of neuropathic pain, as well as other disorders.1b,c
Results
Reaction development
N-Sulfinyl ureas have emerged as promising enantioselective organocatalysts with the sulfinyl group serving as both a chiral directing group and electron withdrawing substituent10a and have proven to be particularly effective for the addition of the acidic thioacetic acid pronucleophile.10b The previously reported N-sulfinyl urea catalyst 310b (see Fig. 2) was used for initial screening in the enantioselective addition of Meldrum's acid to nitroalkenes 1. In cyclopentyl methyl ether (CPME), which has seen increasing use as solvent for large scale industrial applications,13 the addition to trans-β-nitrostyrene 1a proceeded at rt with 95% ee, while the alkyl nitroalkene 1b, which notably leads to the drug Lyrica,12 provided the addition product 2b in 87% ee (eqn 4). With the goal of ultimately applying the method to the synthesis of Lyrica, we focused subsequent optimization studies on substrate 1b.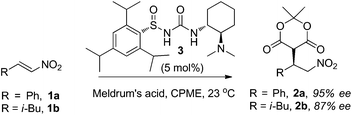 | (4) |
Recent reports for other urea catalysts have shown dramatic differences in the selectivity and activity arising from different tertiary amine moieties.14 On this basis, several new sulfinyl ureas were synthesized and tested in the enantioselective addition of Meldrum's acid to aliphatic nitroalkene 1b (Table 1). Catalysts 4–6, all bearing acyclic tertiary amines, uniformly reduced the enantioselectivity (entries 2–4). Cyclic tertiary amine catalysts, such as pyrrolidine 7 and piperidine 8, showed more promise, with catalyst 8 propelling the reaction to 91% conversion and 92% ee (entry 6). Free amine catalyst 9 exhibited poor solubility and poor conversion (entry 7). Interestingly, diastereomeric catalyst 10 gave drastically reduced selectivity, indicating a substantial matched–mismatched effect arising from the relative configurations of the sulfinyl and 1,2-diamine stereocenters (entry 8). tert-Butanesulfinyl urea 11 was also tested, but was found to be less selective than the corresponding trisyl sulfinyl urea 3 (entry 9).15
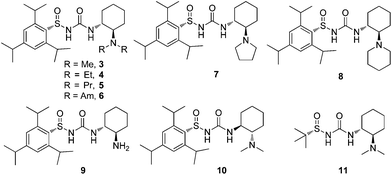 | ||
| Fig. 2 Catalysts tested in the addition of Meldrum's acid to nitroalkenes. | ||
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Conversiona (%) | eeb,c (%) |
| a Conversion was determined by 1H NMR from the ratio of the product to starting material. b Enantiomeric excess was determined by chiral HPLC. c Absolute stereochemistry was determined by the correlation of the optical rotation of product 2b to the literature value.5b | |||
| 1 | 3 | 76 | 87 |
| 2 | 4 | 62 | 80 |
| 3 | 5 | 71 | 77 |
| 4 | 6 | 82 | 81 |
| 5 | 7 | 83 | 91 |
| 6 | 8 | 91 | 92 |
| 7 | 9 | 29 | 70 |
| 8 | 10 | 73 | −64 |
| 9 | 11 | 70 | 79 |

Low catalyst loading was sought to enhance the practical utility of this transformation. However, the reaction rate was limited by the low solubility of Meldrum's acid in CPME and other nonpolar solvents (more polar solvents result in diminished enantioselectivities). We postulated that using a more hydrophobic Meldrum's acid derivative might circumvent this problem. Indeed, cyclohexyl Meldrum's acid exhibited enhanced (∼4-fold at 23 °C) solubility in CPME. The resulting increase in the effective concentration allowed the catalyst loading to be reduced to 1 mol% at room temperature. Additionally, a slight increase in enantioselectivity was observed upon switching to cyclohexyl Meldrum's acid (Table 2, entry 2).
|
|
||||
|---|---|---|---|---|
| Entry | R | Producta | Yieldb (%) | eec (%) |
| a Reactions were performed with 1.0 mol% catalyst loading at 0.3 M concentration of substrate with 1.5 equiv. of cyclohexyl Meldrum's acid. b Isolated yield of analytically pure material after chromatography. c Enantiomeric excess was determined by chiral HPLC. d Reaction was run using 3.0 equiv. of cyclohexyl Meldrum's acid. | ||||
| 1 | C6H5 | 12a | 95 | 98 |
| 2 | i-Bu | 12b | 90 | 94 |
| 3 | o-MeC6H4 | 12c | 94 | 96 |
| 4 | p-MeC6H4 | 12d | 99 | 98 |
| 5 | p-CF3C6H4 | 12e | 82 | 98 |
| 6 | p-MeOC6H4 | 12f | 99 | 96 |
| 7 | o,p-Cl2C6H3 | 12g d | 95 | 96 |
| 8 | n-Pr | 12h | 94 | 94 |
| 9 | c-Hex | 12i | 73 | 96 |
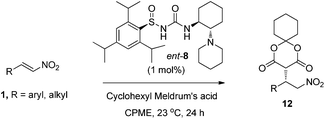
Synthetic scope
The substrate scope was explored for the addition of cyclohexyl Meldrum's acid to various aromatic and aliphatic nitroalkenes 1 using 1 mol% of catalyst ent-8 (the enantiomer of catalyst 8) (Table 2). trans-β-Nitrostyrene 1a underwent addition in 95% yield and 98% ee (entry 1) and the aliphatic Lyrica precursor 1b gave the product 12b in 90% yield and 94% ee (entry 2). Substitution around the aromatic ring was well tolerated, giving up to 99% yield and 98% ee over a range of aromatic substrates (entries 3–7). The enantioselectivity was also excellent for both electron-deficient and -rich derivatives (entries 5 and 6, respectively). 2,4-Dichlorophenyl substrate 1g reacted more slowly, but increasing the amount of cyclohexyl Meldrum's acid afforded the product in 95% yield and 96% ee (entry 7). Moreover, both linear (entry 8) and branched (entries 2 and 9) β-alkyl substituted nitroalkenes provided addition products in good yields and with high selectivities (≥94% ee).Additions to α,β-disubstituted nitroalkenes 13 would generate products 14 (Table 3) with two stereocenters, which are desirable for the preparation of bioactive compounds (e.g., see Fig. 1). However, these more highly substituted nitroalkenes present several additional challenges. Firstly, under basic conditions, the acidity of the proton α to the nitro group of 14 can cause epimerization of that stereocenter, leading to a thermodynamic ratio of diastereomers. Secondly, for acyclic nitroalkane products 14, the thermodynamic product ratio is very modest.16 Finally, the more substituted nitroalkenes 13 were expected to undergo addition at much slower rates. Nevertheless, we were pleased to find that using catalyst ent-8, α,β-disubstituted nitroalkene 13a afforded addition product 14a with almost perfect diastereoselectivity (Table 3, entry 1). An X-ray crystal structure of product 14a revealed that the relative stereochemistry is consistent with delivery of the nucleophile and proton to the same face of the nitroalkene.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | Producta | Yieldb (%) | Crude d.r.c | eed (%) |
| a Reactions were performed with 3.0 mol% catalyst loading at 0.6 M concentration of substrate and with 3.0 equiv. of cyclohexyl Meldrum's acid. b Isolated yield of analytically pure single diastereomers after chromatography. c Crude diastereomeric ratios were determined by 1H NMR. d Enantiomeric excess was determined by chiral HPLC. e Absolute and relative stereochemistry was determined from an X-ray crystal structure. f Reaction was performed using 1.0 mol% catalyst. g Reaction was performed at 0.05 M concentration. h Reaction was performed at 0.3 M concentration using 1.5 equiv. of cyclohexyl Meldrum's acid. | ||||||
| 1 | C6H5 | Me | 14a e | 90 | 97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 :3 |
93 |
| 2 | p-CF3C6H4 | Me | 14b | 71 | 95:5 |
94 |
| 3 | p-MeOC6H4 | Me | 14c | 91 | 96:4 |
92 |
| 4 | p-ClC6H4 | Me | 14d | 77 | 96:4 |
91 |
| 5 | p-MeC6H4 | Me | 14e | 91 | 97:3 |
92 |
| 6 | C6H5 | Bn | 14f | 74 | 98:2 |
90 |
| 7 | C6H5 | n-Bu | 14g | 70 | 99:1 |
83 |
| 8 | –(CH2)4– | –(CH2)4– | 14h f , g | 78 | 87:13 |
91 |
| 9 | –(CH2)3– | –(CH2)3– | 14i f , g | 93 | 98:2 |
97 |
| 10 | –(CH2)5– | –(CH2)5– | 14j f , h | 92 | >99:1 |
98 |
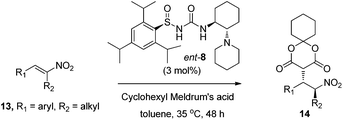
We investigated the substrate scope for the enantioselective addition of cyclohexyl Meldrum's acid to a number of cyclic and acyclic α,β-disubstituted nitroalkenes 13 (Table 3). Despite the lower reactivity of these more challenging substrates, only 3 mol% of catalyst ent-8 was necessary for efficient conversion to addition products 14. Parent substrate 13a afforded the product in 97:3 dr, 93% ee, and after chromatography, a 90% isolated yield of a single diastereomer. Variation of the aromatic ring shows a high tolerance for both electron-rich and electron-poor derivatives, as well as various para-substituents (entries 2–5). The substituent alpha to the nitro group can also be varied from a simple methyl group to other groups such as benzyl (entry 6) and butyl (entry 7) groups. The less challenging cyclic α,β-disubstituted nitroalkenes are also effective substrates, affording products 14h–14j with high selectivities and yields. In fact, these cyclic substrates required only 1 mol% catalyst loading.
Applications to the synthesis of γ-amino acid derivatives
We then sought to demonstrate the utility of these addition products for the construction of biologically relevant γ-amino acid derivatives. Importantly, adduct 14a with two stereocenters can be readily converted to γ-amino acid derivatives2 with retention of the stereochemical information (Scheme 1). The TsOH-catalyzed one-pot hydrolysis/decarboxylation12 of 14a affords monoacid 16 in 93% yield and in a diastereomerically and analytically pure form after simple extractive isolation. Monacid 16 can then be reduced either using Zn dust and HCl to give diastereomerically pure γ-amino acid 172d in 97% yield or by SnCl2 reduction/esterification conditions to provide the amino ethyl ester 18 in 61% overall yield, also in diastereomerically pure form.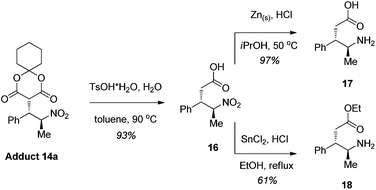 | ||
| Scheme 1 The synthesis of γ-amino acid scaffolds from adduct 14a. | ||
Finally, we sought to demonstrate the utility of our method for drug production with the mole scale synthesis of Lyrica precursor 15 (Scheme 2). Although 1 mol% catalyst loading is sufficient for laboratory scale chemistry, further reduction of the catalyst loading is desirable for large scale drug production. With mild heating to 35 °C, complete conversion can be achieved with only 0.2 mol% of catalyst 8 and with only a very slight reduction in enantioselectivity. The crude addition product can be taken on directly to a one-pot hydrolysis/decarboxylation step, allowing for a telescoped overall process. Key Lyrica intermediate 15 was synthesized by this route on a one mole scale in 90% overall yield from nitroalkene 1b. A one-step conversion of intermediate 15 to Lyrica viahydrogenation has been reported in the literature.12
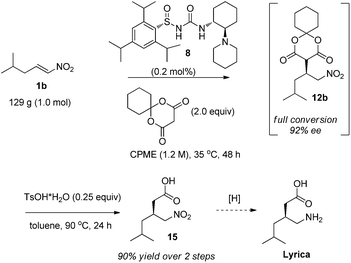 | ||
| Scheme 2 Large-scale production of the key Lyrica intermediate. | ||
Conclusion
In summary, we have introduced a practical method for constructing optically active γ-amino acids that utilizes cyclohexyl Meldrum's acid as a versatile monocarboxylic acid equivalent and piperidinyl sulfinyl urea 8 as a highly selective and efficient organocatalyst. Decarboxylation and nitro reduction can be performed to provide γ-amino acid derivatives without any loss of stereochemistry even for α,β-disubstituted nitroalkene inputs. The viability of this method toward drug production was demonstrated with the mole scale synthesis of Lyrica precursor 15 using only 0.2 mol% of catalyst 8. This method is not only the first example of the highly enantioselective addition of Meldrum's acid derivatives to nitroalkenes, but also provides the first example of the organocatalytic addition of any type of carbon nucleophile to acyclic α,β-disubstituted nitroalkenes to set both the α- and β-stereocenters in acyclic products with high enantio- and diastereoselectivity. The key to obtaining high diastereoselectivity is the acidic nature of Meldrum's acid vs. the more traditional malonate esters. We believe that Meldrum's acid will be similarly advantageous for a variety of transformations that are currently under investigation.Acknowledgements
This work was supported by the NSF (CHE-1049571). We are grateful to Chris Incarvito and the Yale X-ray crystallography for solving the crystal structure of 14a, and to Chris Senanayake and Steve Han for providing trisyl sulfinamide at the early project stage.Notes and references
- For a general review of GABA-related drugs, see: (a) P. Yogeeswari, J. V. Ragavendran and D. Sriram, Recent Pat. CNS Drug Discovery, 2006, 1, 113–118 CrossRef CAS . For reviews of Pregabalin drug activity, see; (b) A. Beydoun, W. Nasreddine and S. Atweh, Expert Rev. Neurother., 2008, 8, 1013–1024 CrossRef CAS; (c) R. B. Silverman, Angew. Chem., Int. Ed., 2008, 47, 3500–3504 CrossRef CAS.
- (a) M. Takagi, K. Ishimitsu, T Nishibe, Preparation of Novel Hetereocyclic Compounds such as 2-Imidazolidinone, 2-Thiazolidinone and 2-Pyrrolidinone Derivatives as Anti-inflammatory Agents, Patent WO 2003031414(A1), 17th April, 2003; (b) J. Cai; X. Hao; Y. Wang; L. Xu; X. Yang; Y Wang. Application of Phenclobenpyrone as Antidepressent. Patent CN 1759831(A), April 19, 2006; (c) J. W. Liebeschuetz, R. B. Katz, A. D. Duriatti and M. L. Arnold, Pestic. Sci., 1997, 50, 258–274 CrossRef CAS; (d) S. Ansorge, U. Bank, K. Nordhoff, M. Tager and F Striggow, Novel Dipeptidyl Peptidase IV Inhibitors used for Functionally Influencing Different Cells and Treating Immunological, Inflammatory, Neuronal and Other Diseases, Patent WO 2005037779(A2), 28th April, 2005; (e) T. R. Belliotti, T. Capiris, I. V. Ekhato, J. J. Kinsora, M. J. Field, T. G. Heffner, L. T. Meltzer, J. B. Schwarz, C. P. Taylor, A. J. Thorpe, M. G. Vartanian, L. D. Wise, Z.-S. Ti, M. L. Weber and D. J. Wustrow, J. Med. Chem., 2005, 48, 2294–2307 CrossRef CAS; (f) I. Kalvins, A. Lebedevs, A. Cernobrovijs, M. Dambrova, L. Zvejniece, M. Vorona and G Veinbergs, 4R,5S–Enantiomer of 2-(5-Methyl-2-Oxo-4-Phenyl-Pyrrolidin-1-yl)-Acetimide with Nootropic Activity, Patent WO 2011045888(A1), 12th May , 2011.
- Organocatalytic enantioselective additions to β-substituted nitroalkenes with high enantioselectivities have been accomplished using malonate esters. For select examples, see: (a) T. Okino, Y. Hoashi and Y. Takemoto, J. Am. Chem. Soc., 2003, 125, 12672–12673 CrossRef CAS; (b) J. Ye, D. J. Dixon and P. S. Hynes, Chem. Commun., 2005, 4481–4483 RSC; (c) H. Li, L. T. Wang and L. Deng, J. Am. Chem. Soc., 2004, 126, 9906–9907 CrossRef CAS; (d) F. Xu, E. Corley, M. Zacuto, D. A. Conlon, B. Pipik, G. Humphrey, J. Murry and D. Tschaen, J. Org. Chem., 2010, 75, 1343–1353 CrossRef CAS; (e) S. H. McCooey and S. J. Connon, Angew. Chem., Int. Ed., 2005, 44, 6367–6370 CrossRef CAS; (f) L. Zhang, M.-M. Lee, S.-M. Lee, J. Lee, M. Cheng, B.-S. Jeong, H.-G. Park and S.-S. Jew, Adv. Synth. Catal., 2009, 351, 3063–3066 CrossRef CAS . For two examples of highly enantioselective metal-catalyzed additions of malonate esters to nitroalkenes, see; (g) D. M. Barnes, J. Ji, M. G. Fickes, M. A. Fitzgerald, S. A. King, H. E. Morton, F. A. Plagge, M. Preskill, S. H. Wagaw, S. J. Wittenberger and J. Zhang, J. Am. Chem. Soc., 2002, 124, 13097–13105 CrossRef CAS; (h) D. A. Evans and D. Seidel, J. Am. Chem. Soc., 2005, 127, 9958–9959 CrossRef CAS.
- (a) See ref. 3a; (b) W.-Y. Chen, P. Li, J.-W. Xie and X.-S. Li, Catal. Commun., 2011, 12, 502–504 CrossRef CAS.
- For examples of Meldrum's acid additions that give low to modest enantioselectivity see: (a) See ref. 3a; (b) O. Bassas, J. Huuskenon, K. Rissanen and A. M. P. Koskinen, Eur. J. Org. Chem., 2009, 1340–1351 CrossRef CAS; (c) E. Kleczkowska and W. Sas, Polish J. Chem., 2007, 81, 1457–1464 CAS.
- Malonates typically require a two-step process of saponification under basic conditions followed by acidification to achieve decarboxylation . For a representative process research example see: M. C. Hillier, J.-F. Marcoux, D. Zhao, E. J. J. Grabowski, A. E. McKeown and R. D. Tillyer, J. Org. Chem., 2005, 70, 8385–8394 CrossRef.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456–463 CrossRef CAS.
- Conjugate addition of diethyl malonate (3.0 equiv.) to trans-β-nitro, β-methyl styrene (1.0 equiv.) in CD2Cl2 using limiting amounts of DBU (1.0 equiv) as the base resulted in the complete consumption of the nitroalkene to afford a 1.8:1 ratio of diastereomers, determined by 1H NMR spectroscopy of the reaction mixture and further confirmed by isolation of the product diastereomers using flash chromatography. In contrast, when cyclohexyl Meldrum's acid was employed under identical conditions, clean conversion (94%) to product with > 97:3 dr was observed in 13 h. Furthermore, attempts to add diethyl malonate to the same alkene, utilizing triethylamine as the base, only afforded ∼5% conversion after 48 h to provide a mixture of diastereomers with low dr. When cyclohexyl Meldrum's acid was used under identical conditions, full conversion (∼24 h) and >20:1 dr was obtained..
- For reviews of H-bonding organocatalysis, see: (a) M. S. Taylor and E. N. Jacobsen, Angew. Chem., Int. Ed., 2006, 45, 1520–1543 CrossRef CAS; (b) Y. Takemoto, Org. Biomol. Chem., 2005, 3, 4299–4306 RSC; (c) A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713–5743 CrossRef CAS; (d) T. Akiyama, Chem. Rev., 2007, 107, 5744–5758 CrossRef CAS; (e) X. Yu and W. Wang, Chem.–Asian J., 2008, 3, 516–532 CrossRef CAS; (f) M. Terada, Synthesis, 2010, 12, 1929–1982 CrossRef; (g) M. Rueping, R. M. Koenigs and I. Atodiresei, Chem.–Eur. J., 2010, 16, 9350–9365 CrossRef CAS.
- For examples of organocatalysts containing sulfinamides, see: (a) M. T. Robak, M. Trincado and J. A. Ellman, J. Am. Chem. Soc., 2007, 129, 15110–15111 CrossRef CAS; (b) K. L. Kimmel, M. T. Robak and J. A. Ellman, J. Am. Chem. Soc., 2009, 131, 8754–8755 CrossRef CAS; (c) H. Xu, S. J. Zuend, M. G. Woll, Y. Tao and E. N. Jacobsen, Science, 2010, 327, 986–990 CrossRef CAS; (d) D. Pei, Z. Wang, S. Wei, Y. Zhang and J. Sun, Org. Lett., 2006, 8, 5913–5915 CrossRef CAS.
- For examples of carbon nucleophile additions to acyclic α,β-disubstituted nitroalkenes, for which cyclization has been relied upon to achieve high diastereoselectivity, see: (a) C.-L. Cao, Y.-Y. Zhou, X.-L. Sun, Y. Tang, Y.-X. Li, G.-Y. Li and J. Sun, Chem.–Eur. J., 2009, 15, 11384–11389 CrossRef CAS; (b) S. Chandrasekhar, K. Mallikarjun, G. Pavankumarreddy, K. V. Rao and B. Jagadeesh, Chem. Commun., 2009, 4985–4987 RSC; (c) M. Rueping, A. Kuenkel and R. Frohlich, Chem.–Eur. J., 2010, 16, 4173–4176 CrossRef CAS; (d) Y. Wang, S. Zhu and D. Ma, Org. Lett., 2011, 13, 1602–1605 CrossRef CAS.
- After one-pot hydrolysis and decarboxylation, the nitro monoacid is obtained. This compound has been carried onto Lyrica, see: V. S. K. Bobba; S. R. Sanikommu, B. M. More, D. S. Metil, P. B. Kulkarni, K. J. Prabahar, M. Khan, Novel Process for Preparing Pregabalin and its Acid Addition Salts, U.S. Patent WO 2008117305(A2), 2nd October, 2008.
- For leading references on CPME, see: (a) V. Antonucci, J. Coleman, J. B. Ferry, N. Johnson, M. Mathe, J. P. Scott and J. Xu, Org. Process Res. Dev., 2011 Search PubMed , ASAP; (b) K. Watanabe, N. Yamagiwa and Y. Torisawa, Org. Process Res. Dev., 2007, 11, 251–258 CrossRef CAS.
- (a) D. E. Fuerst and E. N. Jacobsen, J. Am. Chem. Soc., 2005, 127, 8964–8965 CrossRef CAS; (b) A. Berkessel, F. Cleemann, S. Mukherjee, T. N. Mueller and J. Lex, Angew. Chem., Int. Ed., 2005, 44, 807–811 CrossRef CAS; (c) A. Berkessel, S. Mukherjee, T. N. Mueller, F. Cleemann, K. Roland, M. Brandenburg, J.-M. Neudoerfl and J. Lex, Org. Biomol. Chem., 2006, 4, 4319–4330 RSC; (d) G. E. Veitch and E. N. Jacobsen, Angew. Chem., Int. Ed., 2010, 49, 7332–7335 CrossRef CAS; (e) L. Li, E. G. Klauber and D. J. Seidel, J. Am. Chem. Soc., 2008, 130, 12248–12249 CrossRef CAS.
- For the development and applications of triisopropylphenyl sulfinamide, see: Z. Han, D. Krishnamurthy, P. Grover, Q. K. Fang, D. A. Pflum and C. H. Senanayake, Tetrahedron Lett., 2003, 44, 4195–4197 CrossRef CAS.
-
(a) The thermodynamic ratio of diastereomers 14a was found to be 1.3:1. The ratio was determined by 1H NMR after the α-nitro stereocenter of 14a had been epimerized in the presence of excess triethylamine at 60 °C;
(b) For an example of addition to acyclic α,β-disubstituted nitroalkenes in which low dr was observed, see: M. Ganesh and I. N. N. Namboothiri, Tetrahedron, 2007, 63, 11973–11983 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC reference numbers 833178. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00441g |
| This journal is © The Royal Society of Chemistry 2012 |