A solid solution approach to 2D coordination polymers for CH4/CO2 and CH4/C2H6 gas separation: equilibrium and kinetic studies†‡
Satoshi
Horike
a,
Yasutaka
Inubushi
b,
Takashi
Hori
b,
Tomohiro
Fukushima
a and
Susumu
Kitagawa
*acd
aDepartment of Synthetic Chemistry and Biological Chemistry, Graduate School of Engineering, Kyoto University, Katsura, Nishikyo-ku, Kyoto 615-8510, Japan. E-mail: kitagawa@icems.kyoto-u.ac.jp; Fax: +81-75-383-2732; Tel: +81-75-383-2736
bSynthesis Research Laboratory, Kurashiki Research Center, Kuraray Co. Ltd., 2045-1, Sakazu, Kurashiki, Okayama 710-0801, Japan
cInstitute for Integrated Cell-Material Sciences (iCeMS), Kyoto University, Yoshida, Sakyo-ku, Kyoto 606-8501, Japan
dERATO Kitagawa Integrated Pores Project, Japan Science and Technology Agency (JST), Kyoto Research Park, Shimogyo-ku, Kyoto 600-8815, Japan
First published on 6th October 2011
Abstract
Gas separation properties for CH4/CO2 and CH4/C2H6 of flexible 2D porous coordination polymers under equilibrium gas conditions and mixed gas flow conditions were investigated and the gas separation efficiencies were optimized by precise tuning of the flexibility in ligand-base solid solution compounds.
Introduction
The development of materials for gas separation technology has become increasingly important in recent times.1 For example, the separation of CO2 from CH4/CO2 mixtures in biogas and C2H6 from CH4/C2H6 mixtures in natural gas exhausts is facilitated by the use of solid adsorbents. The pressure swing adsorption (PSA) process and the use of solid support membranes are well-known technologies and in both cases, the selective binding of the target gas and gas adsorption kinetics are important factors for the design of the adsorbents.2,3 In particular, the conditions used in the PSA process depend on the target gas produced by various industrial processes and therefore porous materials that have the appropriate separation properties for specific working conditions have been required.4 Porous coordination polymers (PCPs) or metal–organic frameworks (MOFs) consist of metal ions and organic ligands that are promising candidates for CO2 gas separation.5–20 The pore surface can be designed to be either basic or acidic and the pore dimensions and diameter are readily tunable, ranging from 0.3 nm to a few nm. Some compounds that exhibit a “flexible” nature in the framework have been highlighted for gas separation.21–26 The flexibility affords a structure transformation from a non-porous to a porous system via gas adsorption where the transformation depends on the gaseous species. This phenomenon often results in gas separation (or recognition) properties. Indeed, we have succeeded in controlling flexibility by employing a solid solution where two distinct PCP/MOFs are uniformly integrated into a single framework with an arbitrary ratio.27 Considering the potential use of these materials in separation processes, we have to evaluate them not only under equilibrium conditions but also under kinetic dynamic conditions at operating temperatures and pressures employed in gas separation. As PSA, for instance, typically runs under ambient temperature and a total pressure of 0.4 ∼ 0.8 MPa, the cycle time is several minutes. This process requires a quick separation ability. In this work, we evaluated CO2 and C2H6 gas separation from CH4 under both equilibrium and kinetic conditions using flexible 2D layer-type PCP/MOFs and optimized their selectivity performance. Fine tuning of the structure flexibility in the frameworks turned out to be a significant factor in the design of materials for potential application in PSA processes for CH4/CO2 and CH4/C2H6 gas mixtures.Results and discussion
We employed two PCP/MOF frameworks, [Zn(5NO2-ip)(bpy)]n (CID-5, 5NO2-ip = 5-nitroisophthalate, bpy = 4,4′-bipyridyl) and [Zn(5MeO-ip)(bpy)]n (CID-6, 5MeO-ip = 5-methoxyisophthalate), both of which possess microporous 2D layer stacking structures.27 These can be regarded as a series of porous coordination polymers with interdigitated structures, which we denote as CIDs.27–34 We also fabricated three ligand-base solid solution compounds of CID-5 and CID-6. The formula is depicted as [Zn(5NO2-ip)1−x(5MeO-ip)x(bpy)]n (CID-5/6), where x is the ratio of 5MeO-ip ligands in the structures. In the crystal structures, two Zn2+ ions are connected by carboxylate groups and the axial position of the Zn2+ ions are coordinated to the nitrogen atoms of the bpy ligands to form an octahedral coordination environment as shown in Fig. 1. Dicarboxylates and Zn2+ form ribbon-type infinite 1D chains and each chain is linked by bpy to create 2D layers. The layers are stacked to complete the interdigitated porous structure. It has been reported that the structure flexibility of CID-5 is larger than that of CID-6.27CID-5 has a large flexibility that results in a significant structure transformation from totally non-porous to porous. CID-6 is regarded as a rigid (robust) type porous framework where its porosity hardly changes during guest containment or the removal phase. The origin of their flexibility could be caused by the size/shape and electron-donating/-withdrawing properties of the substituent group of ligands and the resulting packed structures. We also confirmed that the three CID-5/6 (x = 0.1, 0.2, 0.4) were all single-phase materials by powder X-ray diffraction (XRD) studies and elemental analysis. This means that the two different ligands (5NO2-ip and 5MeO-ip) are evenly distributed in the crystals regardless of the ratio of the ligands. CID-5/6 (x = 0.1) has characteristics similar to those of pure CID-5 with regard to crystal structure. However, CID-5/6 (x = 0.2, 0.4) compounds are more closely related to pure CID-6 and their flexibility depends on the x content; as x increases, the flexibility decreases.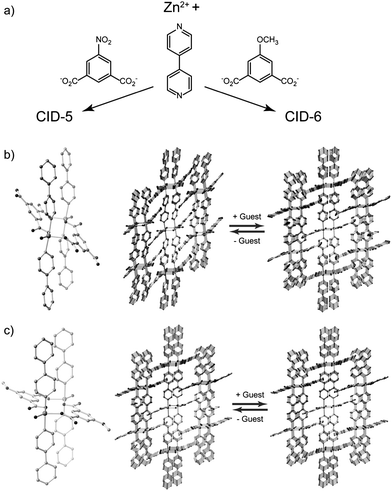 | ||
| Fig. 1 (a) Reaction schemes of CID-5 and CID-6. Crystal structures of the coordination environment around the Zn2+ ions and assembled 2D layer stacking of (b) CID-5 and (c) CID-6 before and after guest adsorption. Guests are omitted. | ||
We then measured CO2 and CH4 gas adsorption isotherms for five CID compounds at 273 K as shown in Fig. 2. For CID-5, the total uptake of CO2 was 57 mL g−1, where the profile is not a typical Type-I isotherm according to the IUPAC classification but a gate-opening-type adsorption.29,35–39 Adsorption does not occur until the pressure reaches a specific point (in this case 0.10 MPa), upon which it starts abruptly and then reaches saturation. CID-5 adsorbs less than 5 mL g−1 of CH4 at 1.0 MPa and 273 K, indicating that structure transformation does not occur at this point. Interaction between CH4 and the framework was not sufficient to open the structure under the measurement conditions. On the other hand, CID-6 has permanent porosity in the deguested form and shows a Type-I adsorption isotherm in which gas uptake starts at the low-pressure region. Because of permanent microporosity, CID-6 adsorbs both CO2 and CH4, and the total uptake at 1.0 MPa and 273 K is 58 mL g−1 (CO2) and 45 mL g−1 (CH4), respectively. We observed distinct adsorption behaviour on CID-5 and CID-6 that can be attributed to structure flexibility. Gas adsorption isotherms of CID-5/6 and the ligand-base solid solution of CID-5 and CID-6 are also shown in Fig. 2c–2e. As discussed before, the structure characteristics of CID-5/6 such as porosity and flexibility depend on the relative ratio of each ligand (x) in the framework. Regarding CO2, CID-5/6 (x = 0.2, 0.4) showed Type-I isotherms that were similar to CID-6 because they have permanent porosity in the deguested form. Meanwhile, CID-5/6 (x = 0.1) was dominated by the flexible properties of CID-5, and the CO2 isotherm demonstrated gate-opening-type behaviour, even though the gate-opening pressure was 0.02 MPa, which was smaller than that for pure CID-5. The reason for the lowering of the gate-opening pressure is that the thermodynamic stability of the deguested porous framework of CID-5/6 (x = 0.1) was lower than pure CID-5 because of the partial doping of the CID-6 domain. For CH4, the adsorption isotherms were more sensitive to the ligand ratio in CID-5/6. As the content of 5MeO-ip ligand (x) decreases, the adsorption profiles became more gradual and the total amount of adsorption was low. Although the adsorption isotherms of CID-5/6 (x = 0.2, 0.4) were Type-I, the isotherm of CID-5/6 (x = 0.1) was not Type-I but appeared to be a Type-III isotherm35 because the compound was more closely related to pure CID-5 with high flexibility and a non-porous structure. Because of the decrease of cooperativity in the framework of CID-5/6 (x = 0.1) for gate-opening behaviour, the structure transformation from non-porous to porous became gradual, which resulted in the apparent Type-III isotherm. As a result, we observed a strong dependency of the gas adsorption isotherms for CO2 and CH4 in the ambient temperature and pressure region as the ratio of ligand in the ligand-base solid-solution-type CID compounds was changed.
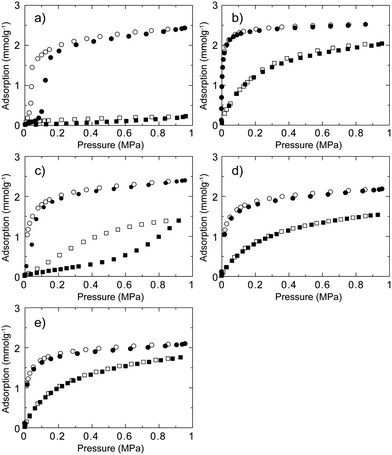 | ||
| Fig. 2 CH4 (square) and CO2 (circle) sorption isotherms for (a) CID-5, (b) CID-6, (c) CID-5/6 (x = 0.1), (d) CID-5/6 (x = 0.2) and (e) CID-5/6 (x = 0.4) at 273 K. Adsorption: closed characters; desorption: open characters. | ||
We also studied the gas separation properties under kinetic gas flow conditions at ambient conditions. The breakthrough curve is a typical way of evaluating the gas separation ability for adsorbents under flowing gas conditions that are related to the PSA process. We measured the gas separation properties of CID-5, 6, CID-5/6 (x = 0.1, 0.4) (Fig. 3) for the CH4/CO2 mixture at 273 K and a total pressure of 0.80 MPa. The breakthrough curve of CID-5 is shown in Fig. 3a. After injection of the gas mixture, the concentration of CO2 detected by gas chromatography (GC) was 10% and 90% for CH4 and the retention time was around 10 min. Then it reached the breakpoint and smoothly went back to the initial ratio of the gas mixture. Considering that the relative pressure of CO2 (0.32 MPa) in the gas mixture was higher than the gate-opening pressure of CID-5 (0.10 MPa) studied by an equilibrium isotherm, the compound adsorbed CO2 and was accompanied by a structure transformation. On the other hand, the relative pressure of CH4 was 0.48 MPa and CID-5 did not show uptake of gas at this pressure and consequently we observed a concentration effect of CO2 over CH4 from the flowing gas. Even though we observed a concentration effect of the target gas under flowing conditions, the detected concentration of CH4 was 90% and not close to 100%, which is not sufficient for purification of the target gas. A plausible mechanism for this behaviour is the following: as the CH4/CO2 gas mixture reaches the powder of CID-5 at the first point of adsorption, it starts to selectively adsorb CO2 with a gate-opening phenomenon. Then, the relative pressure of CO2 in the column of CID-5 decreases quickly, with the result that the relative pressure is now below the gate-opening pressure. The gas mixture of CH4/CO2 in which the relative pressure of CO2 detected at the outlet is below the gate-opening pressure caused the detection ofca. 10% of CO2 before the breakpoint was reached.
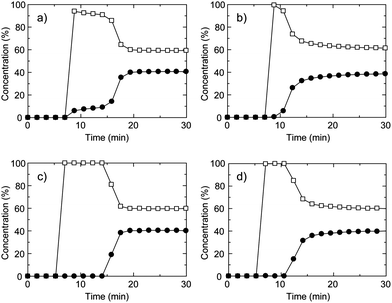 | ||
Fig. 3 Breakthrough curves of CH4/CO2 mixture (60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 (vol)) for (a) CID-5, (b) CID-6, (c) CID-5/6 (x = 0.1) and (d) CID-5/6 (x = 0.4). The open square is CH4 and the closed circle is CO2. These were measured at 273 K, the total pressure was 0.80 MPa and the space velocity was 6 min−1. :40 (vol)) for (a) CID-5, (b) CID-6, (c) CID-5/6 (x = 0.1) and (d) CID-5/6 (x = 0.4). The open square is CH4 and the closed circle is CO2. These were measured at 273 K, the total pressure was 0.80 MPa and the space velocity was 6 min−1. | ||
The breakthrough curve of CID-6 had a short retention time for separation of CO2 and CH4 even though it possesses permanent microporosity in the deguested form (Fig. 3b). It reached the breakthrough point in minutes and then returned to the initial gas ratio where negligible separation was observed. This is because CID-6 adsorbs both CO2 and CH4 with a Type-I isotherm and under kinetic gas flow conditions, co-adsorption of both gases occurred and no clear separation from the breakthrough curve was observed. This indicates that CID-6 could not effectively capture CO2 over CH4 under the investigated gas flow conditions because of the absence of specific binding sites or flexibility for selective CO2 adsorption in the framework.
To improve the separation properties, we tried optimizing the performance with a solid solution of CID-5/6 (x = 0.1, 0.4). As shown in Fig. 3d, CID-5/6 (x = 0.4) having similar properties to pure CID-6 demonstrated improved separation behaviour because it has more favourable characteristics for CO2 capture. The gas detected first by GC was CH4 only, with no detection of CO2 indicating high selectivity for CO2 over CH4 under flowing conditions. After a few minutes, it reached the breakpoint and then returned to the original gas ratio. CID-5/6 (x = 0.1), whose structure is close to pure CID-5 but doped with a small amount of CID-6, demonstrated improved separation properties (Fig. 3c). The gate-opening pressure of CO2 for CID-5/6 (x = 0.1) was 0.04 MPa, which was lower than the relative pressure of CO2 in the gas mixture. These characteristics afforded an appropriate condition for breakthrough measurement of CH4/CO2 and it selectively adsorbed CO2 over CH4 with a retention time of 8 min. In this period, there was no detection of CO2 through the column and almost 100% selectivity of CO2 over CH4 was achieved. Consequently, the ligand-base solid solutions of a CID framework made it possible to tune the gate-opening pressure, which contributed to the optimization of the gas separation properties under both equilibrium and kinetic adsorption conditions.
Not only is CO2 separation from a CH4/CO2 mixture of interest, but so also is separation of C2H6 from a CH4/C2H6 mixture because of the potential application in the separation of C2H6 from natural gas.40C2H6 is subsequently applicable for conversion to various C2 and C3 chemicals. A physical property such as the boiling point of C2H6 (184 K) is similar to CO2 (195 K) and we investigated the separation performance of CID compounds for CH4/C2H6 separation. Gas adsorption and desorption isotherms of C2H6 for CID-5, 6, 5/6 (x = 0.1, 0.2, 0.4) at 273 K are shown in Fig. 4. Similar to CO2, CID-5 afforded gate-opening-type adsorption with a gate-opening pressure of 0.13 MPa, which was slightly higher than that for CO2. The saturated amount of uptake was 59 mL g−1. The gate-opening pressures of CID-5 for CO2 and C2H6 were similar because the adsorbent–adsorbate interactions were similar in both cases. CID-6 demonstrated a Type-I isotherm and adsorbed 50 mL g−1 of C2H6 at 0.95 MPa. Adsorption behaviours of the ligand-base solid solutions, CID-5/6, were also similar to CO2. CID-5/6 (x = 0.2, 0.4) had Type-I isotherms because the crystal structure and flexibility were comparable with those of pure CID-6. CID-5/6 (x = 0.1) demonstrated gate-opening type behaviour even though the pressure point was very low (0.04 MPa). Given that the CH4 adsorption isotherm of CID-5/6 (x = 0.1) was Type-III, the apparent separation of CH4 and C2H6 was investigated under ambient equilibrium conditions.
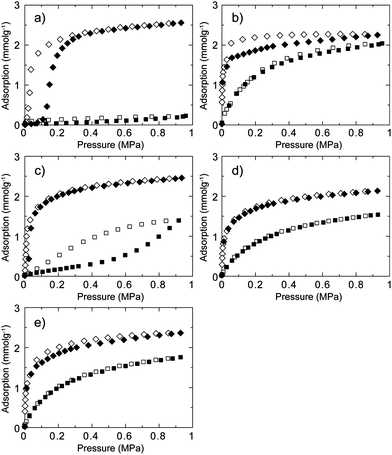 | ||
| Fig. 4 CH4 (square) and C2H6 (diamond) sorption isotherms for (a) CID-5, (b) CID-6, (c) CID-5/6 (x = 0.1), (d) CID-5/6 (x = 0.2) and (e) CID-5/6 (x = 0.4) at 273 K. Adsorption: closed characters; desorption: open characters. | ||
To investigate the separation of C2H6 over CH4 under gas flow conditions, we measured breakthrough curves for a gas mixture of C2H6 and CH4 at 273 K (Fig. 5). The relative pressures of C2H6 in natural gas exhausts are variable depending on the situation and we set a low relative pressure of C2H6 to evaluate the separation ability over CH4. The relative pressure of C2H6 is 0.07 MPa, which is smaller than that for CO2 gas in the CH4/CO2 mixture gas studied in above. As shown in Fig. 5a, the breakthrough curve for CID-5 showed negligible separation properties and quickly reached the breakpoint without any retention time. This behaviour is different from the case of CH4/CO2. This is because the relative pressure of C2H6 (0.07 MPa) was smaller than the gate-opening pressure (0.13 MPa) determined by the equilibrium adsorption measurement. The small fraction of C2H6 in the flowing gas was not able to promote the structure transformation of CID-5 from non-porous to porous. CID-6 also did not demonstrate separation of C2H6 because of co-adsorption of CO2 and C2H6 in the low-pressure region and the obtained breakthrough curve was similar to CID-5. As a result, the two pure CID frameworks did not demonstrate appropriate separation properties under the gas flow conditions. We also investigated the solid solution compounds. CID-5/6 (x = 0.4) in Fig. 5d afforded an almost identical breakthrough curve compared with CID-6 because it has similar characteristics to CID-6, and we also did not observe an efficient retention time for separation. On the contrary, the profile for CID-5/6 (x = 0.1) was obviously different from the other three breakthrough curves: 100% selectivity of C2H6 over CH4 was observed and the retention time reached 25 min under the measurement conditions. Although the volumetric percentage of C2H6 in the gas mixture was only 10%, CID-5/6 (x = 0.1) could selectively adsorb C2H6. The retention time was sufficient for the PSA process and we succeeded in optimizing the separation performance by the solid solution approach. With the systematic control of the intrinsic flexibility of soft PCP/MOFs, we can design various adsorbents for separation under specific conditions.
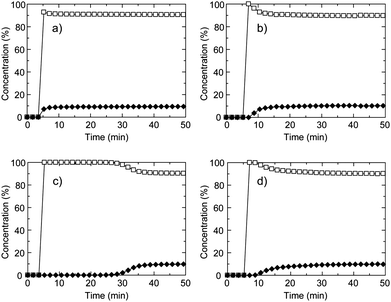 | ||
| Fig. 5 Breakthrough curves of CH4/C2H6 mixture (90:10 (vol)) for (a) CID-5, (b) CID-6, (c) CID-5/6 (x = 0.1) and (d) CID-5/6 (x = 0.4). The open square is CH4 and the closed diamond is C2H6.These were measured at 273 K, the total pressure was 0.80 MPa and the space velocity was 6 min−1. | ||
Conclusions
The separations of gases such as CO2 or C2H6 from biogas or natural gas have so far been significant challenges and will remain so in the near future. The design of porous adsorbents that show high separation performance with low energy consumption is required for purification of the target gas. In this work, we evaluated the CO2 and C2H6 gas separation properties of flexible type 2D PCP/MOF compounds under equilibrium and gas flow conditions. Equilibrium gas sorption isotherms and kinetic breakthrough curves indicated a significant contribution from the flexibility of porous frameworks. To improve gas separation under kinetic conditions, we succeeded in employing the solid solution approach for precise tuning of the gate-opening pressure. We observed improved gas separation properties for both equilibrium and kinetic conditions. The flexible frameworks also have the advantage that they could release adsorbed gas by a moderate treatment such as a gas purging procedure and this property is under investigation. The solid solution synthesis is achievable even on the large scale via a one-pot reaction and the optimization of adsorbents could contribute to the development of separation technologies including the PSA process for various target gases.Notes and references
-
R. T. Yang, Gas Separation by Adsorption Processes, 1997, Imperial College Press Search PubMed
.
- V. G. Gomes and K. W. K. Yee, Sep. Purif. Technol., 2002, 28, 161–171 CrossRef CAS
- L. M. Robeson, J. Membr. Sci., 2008, 320, 390–400 CrossRef CAS
- T. Ren, M. Patel and K. Blok, Energy, 2006, 31, 425–451 CrossRef CAS
- M. Eddaoudi, D. B. Moler, H. L. Li, B. L. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319–330 CrossRef CAS
- B. Kesanli and W. B. Lin, Coord. Chem. Rev., 2003, 246, 305–326 CrossRef CAS
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS
- R. Q. Snurr, J. T. Hupp and S. T. Nguyen, AIChE J., 2004, 50, 1090–1095 CrossRef CAS
- D. Bradshaw, J. B. Claridge, E. J. Cussen, T. J. Prior and M. J. Rosseinsky, Acc. Chem. Res., 2005, 38, 273–282 CrossRef CAS
- L. Pan, B. Parker, X. Y. Huang, D. H. Olson, J. Lee and J. Li, J. Am. Chem. Soc., 2006, 128, 4180–4181 CrossRef CAS
- G. Férey, Chem. Soc. Rev., 2008, 37, 191–214 RSC
- R. E. Morris and P. S. Wheatley, Angew. Chem., Int. Ed., 2008, 47, 4966–4981 CrossRef CAS
- A. Demessence, D. M. D'Alessandro, M. L. Foo and J. R. Long, J. Am. Chem. Soc., 2009, 131, 8784–8786 CrossRef CAS
- J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC
- B. L. Chen, S. C. Xiang and G. D. Qian, Acc. Chem. Res., 2010, 43, 1115–1124 CrossRef CAS
- J. T. Hupp and O. K. Farha, Acc. Chem. Res., 2010, 43, 1166–1175 CrossRef
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef CAS
- C. Gucuyener, J. van den Bergh, J. Gascon and F. Kapteijn, J. Am. Chem. Soc., 2010, 132, 17704–17706 CrossRef
- H. Bux, C. Chmelik, R. Krishna and J. Caro, J. Membr. Sci., 2011, 369, 284–289 CrossRef CAS
- R. Krishna and J. M. van Baten, Phys. Chem. Chem. Phys., 2011, 13, 10593–10616 RSC
- R. Kitaura, K. Seki, G. Akiyama and S. Kitagawa, Angew. Chem., Int. Ed., 2003, 42, 428–431 CrossRef CAS
- P. L. Llewellyn, S. Bourrelly, C. Serre, Y. Filinchuk and G. Ferey, Angew. Chem., Int. Ed., 2006, 45, 7751–7754 CrossRef CAS
- B. L. Chen, C. D. Liang, J. Yang, D. S. Contreras, Y. L. Clancy, E. B. Lobkovsky, O. M. Yaghi and S. Dai, Angew. Chem., Int. Ed., 2006, 45, 1390–1393 CrossRef CAS
- S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695–704 CrossRef CAS
- M. P. Suh and H. S. Choi, Angew. Chem., Int. Ed., 2009, 48, 6865–6869 CrossRef
- G. Férey and C. Serre, Chem. Soc. Rev., 2009, 38, 1380–1399 RSC
- T. Fukushima, S. Horike, Y. Inubushi, K. Nakagawa, Y. Kubota, M. Takata and S. Kitagawa, Angew. Chem. Int. Ed., 2010, 49, 4820–4824 CAS
- S. Horike, D. Tanaka, K. Nakagawa and S. Kitagawa, Chem. Commun., 2007, 3395–3397 RSC
- D. Tanaka, K. Nakagawa, M. Higuchi, S. Horike, Y. Kubota, T. C. Kobayashi, M. Takata and S. Kitagawa, Angew. Chem., Int. Ed., 2008, 47, 3914–3918 CrossRef CAS
- K. Nakagawa, D. Tanaka, S. Horike, S. Shimomura, M. Higuchi and S. Kitagawa, Chem. Commun., 2010, 46, 4258–4260 RSC
- D. Tanaka, A. Henke, K. Albrecht, M. Moeller, K. Nakagawa, S. Kitagawa and J. Groll, Nat. Chem., 2010, 2, 410–416 CrossRef CAS
- H. Sato, R. Matsuda, K. Sugimoto, M. Takata and S. Kitagawa, Nat. Mater., 2010, 9, 661–666 CrossRef CAS
- Y. Hijikata, S. Horike, M. Sugimoto, H. Sato, R. Matsuda and S. Kitagawa, Chem.–Eur. J., 2011, 17, 5138–5144 CrossRef CAS
- Y. Hijikata, S. Horike, D. Tanaka, J. Groll, M. Mizuno, J. Kim, M. Takata and S. Kitagawa, Chem. Commun., 2011, 47, 7632–7634 RSC
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS
- F. X. Coudert, M. Jeffroy, A. H. Fuchs, A. Boutin and C. Mellot-Draznieks, J. Am. Chem. Soc., 2008, 130, 14294–14302 CrossRef CAS
- H. J. Choi, M. Dincă and J. R. Long, J. Am. Chem. Soc., 2008, 130, 7848–7850 CrossRef CAS
- H. Chun and J. Seo, Inorg. Chem., 2009, 48, 9980–9982 CrossRef CAS
- A. Kondo, H. Kajiro, H. Noguchi, L. Carlucci, D. M. Proserpio, G. Ciani, K. Kato, M. Takata, H. Seki, M. Sakamoto, Y. Hattori, F. Okino, K. Maeda, T. Ohba, K. Kaneko and H. Kanoh, J. Am. Chem. Soc., 2011, 133, 10512–10522 CrossRef CAS
- R. W. Baker, Ind. Eng. Chem. Res., 2002, 41, 1393–1411 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: synthesis of compounds. See DOI: 10.1039/c1sc00591j |
| ‡ This work was supported by New Energy and Industrial Technology Development Organization (NEDO) and Grants-in-Aid for Scientific Research, Japan Society for the Promotion of Science (JSPS). |
| This journal is © The Royal Society of Chemistry 2012 |