Postsynthetic ligand exchange as a route to functionalization of ‘inert’ metal–organic frameworks†
Min
Kim‡
a,
John F.
Cahill‡
a,
Yongxuan
Su
a,
Kimberly A.
Prather
*ab and
Seth M.
Cohen
*a
aDepartment of Chemistry and Biochemistry, University of California, San Diego, La Jolla, California 92093, USA. E-mail: scohen@ucsd.edu; Fax: +1-858-822-5598; Tel: +1-858-822-5596
bScripps Institute of Oceanography, University of California, San Diego, La Jolla, California 92093, USA
First published on 13th September 2011
Abstract
Herein, we report that the exchange of ligands from an intact metal–organic framework (MOF) can be exploited as a means to introduce functionalized ligands into MOFs under mild conditions. It is shown that ligand exchange can occur with ‘inert’ Zr(IV)-based UiO-66 MOFs in a solvent dependent manner. We call this process postsynthetic exchange (PSE) and show that it provides access to MOFs that are not readily prepared in high quality by solvothermal methods. It was found that ligand exchange can occur between UiO-66 MOFs as monitored by aerosol time-of-flight mass spectrometry (ATOFMS). ATOFMS was used to analyze the chemical composition of microcrystalline MOFs on the single particle level, providing information not available through bulk analysis. PSE is an important postsynthetic approach to the modification of MOFs, and the ligand exchange revealed by ATOFMS requires a re-evaluation of the assumed ‘stability’ of even the most robust MOFs.
Introduction
Metal–organic frameworks (MOFs) are hybrid materials that are constructed from the assembly of organic ligands and metal ions or metal ion clusters. MOFs are highly porous, crystalline solids that have garnered substantial attention for use in gas adsorption,1 separations,2 and catalysis.3,4 MOFs have attractive features when compared to other crystalline solids including exceptional chemical tunability, high porosity, and good thermal stability. Recent advancements have focused on making MOFs that are more chemically stable, particularly with respect to water, which would be present in many of the industrial applications for which MOFs may be used. Water-stable MOFs have been achieved by a variety of strategies, including the use of more oxophilic, high oxidation state metal ions (e.g.Zr(IV) in UiO-66),5 incorporation of kinetically inert metal ions (e.g.Cr(III) in MIL-101),6 or by changing both the metals and ligands to obtain more robust compositions (e.g. ZIFs).7 Most of the aforementioned materials are obtained largely as microcrystalline powders (with some exceptions, see Behrens et al.)8 that have been reported to be stable in and unaffected by water and other polar solvents (even in the presence of acids) based on powder X-ray diffraction (PXRD) data. Despite their structural robustness, herein we report ligand exchange between MOFs in both particle-to-particle and solution-to-particle fashions and show that ligand exchange can be readily achieved on an inert MOF. Using derivatives of the Zr(IV)-based UiO-66 microcrystalline MOF, we used aerosol time-of-flight mass spectrometry (ATOFMS)9 to uncover an otherwise unknown ligand exchange process that occurs in polar solvents, including water. This exchange process occurs without any apparent loss in crystallinity or structural fidelity of the material, as gauged by PXRD and gas sorption experiments. These findings demonstrate that ATOFMS is a powerful tool for the analysis of microcrystalline materials revealing an underlying lability in an otherwise inert framework material, and thereby has significant implications for how MOF stability is defined and verified. Furthermore, the realization that even highly stable MOFs undergo ligand exchange under select solvent conditions led us to exploit this exchange process for the preparation of unprecedented, functionalized MOFs. In a process termed ‘postsynthetic exchange’ (PSE), unfunctionalized ligands in a MOF crystallite can be readily replaced with highly functionalized ligands without the loss of MOF crystallinity or porosity.10–12 This process is analogous and complimentary to a previous report showing metal ion exchange in MOFs is possible in a single-crystal-to-single crystal (SCSC) fashion.13 Conceptually, PSE has some resemblance to dynamic, covalent scrambling reported in porous organic molecular frameworks.14 Recently, Choe and co-workers published an example of PSE by replacing bipyridine-based ligands within a MOF.15 They confirmed complete replacement of these ligands in a SCSC manner by X-ray structure determination. Precedent for the findings of Choe was provided by several earlier studies that demonstrated the lability of pyridine-based ligands in metal–organic structures.16,17 The carboxylate exchange process described here is perhaps a more surprising finding, due to the greater stability and inertness of the Zr(IV)–carboxylate bond, but is consistent with reports of carboxylate-capped Fe(III) and Zr(IV) clusters being used as synthons for Zr(IV)-based MOFs,18,19 as well as carboxylate exchange in Cu(II)-based metal–organic polyhedra.20 We demonstrate that PSE provides a powerful postsynthetic method for the functionalization of MOFs and thereby the realization of novel MOF materials under mild synthetic conditions.Results and discussion
ATOFMS on microcrystalline MOFs
UiO-66 (UiO = University of Oslo) MOFs were examined because they are formed as microcrystalline powders and are reported to be thermally and chemically stable/inert.21 Specifically, UiO-66-NH2 and UiO-66-Br were utilized due to their distinct chemical compositions that can be readily distinguished by ATOFMS. UiO-66-NH2 and UiO-66-Br were synthesized under solvothermal conditions by combining ZrCl4 with either 2-amino-1,4-benzenedicarboxylate (NH2-BDC) or 2-bromo-1,4-benzenedicarboxylate (Br-BDC), as previously reported (Fig. 1).22UiO-66-NH2 and UiO-66-Br have identical topologies and similar physical characteristics as confirmed by using PXRD, Brunauer–Emmett–Teller (BET) surface area measurements, and thermogravimetric analysis (TGA).22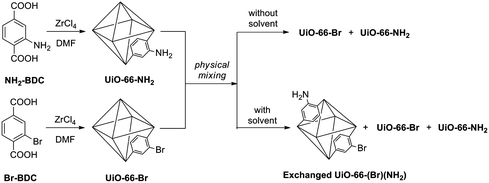 | ||
| Fig. 1 Scheme for the synthesis of UiO-66-NH2 and UiO-66-Br (left) and ligand exchange conditions (right). | ||
ATOFMS was used to differentiate between microcrystalline particles of the two MOF materials. ATOFMS provides single particle size and composition information that has been widely applied in environmental,23 health,24 and biological research.25 Recently, we showed that ATOFMS could be used to confirm the presence of two different ligands in a single particle of microcrystalline UiO-66.26 An aerosol of UiO-66-NH2 and UiO-66-Br was readily produced by sonication of dried samples, and a dilution chamber was used to control the aerosol-flow of UiO particles into the mass spectrometer. Each individual particle mass spectrum was analyzed for the presence of zirconium, bromine, and nitrogen. In the positive ion mass spectra, zirconium produced strong signals at m/z 90–94 and 106–108 for Zr+ and ZrO+, respectively. As expected, the zirconium peaks were observed in all particles of both UiO-66-NH2 and UiO-66-Br. In the negative ion mass spectra, UiO-66-NH2 shows a fragment ion containing carbon and nitrogen at m/z = −26, while UiO-66-Br displays the bromine isotope fragment at m/z = −79 and −81 (Fig. 2 and Fig. S1–S2†).26
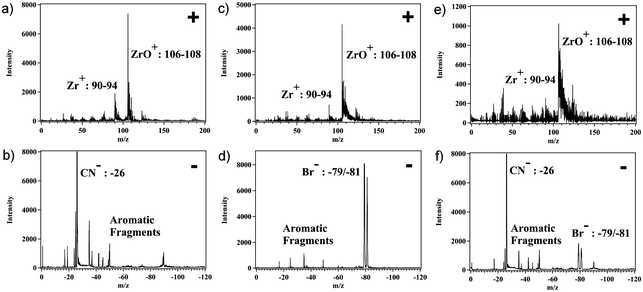 | ||
| Fig. 2 Representative ATOFMS positive ion (a) and negative ion (b) mass spectra of UiO-66-NH2 particles; positive ion (c) and negative ion (d) mass spectra of UiO-66-Br particles; positive ion (e) and negative ion (f) mass spectra of exchanged UiO-66-(Br)(NH2). | ||
Ligand exchange between UiO-66-NH2 and UiO-66-Br
With ATOFMS available as a tool to examine the chemical composition of MOFs at the single-particle level, we sought to examine the chemical stability of these particles. When solid samples of UiO-66-NH2 and UiO-66-Br were physically mixed in the absence of solvent (Fig. 1), no ligand exchange was observed by ATOFMS. That is, the mass spectrum of any individual particle showed either the presence of Br or CN (as found with the parent MOFs), but not both negative ions (Fig. 2, S1 and S2†). However, a mixture of UiO-66-NH2 and UiO-66-Br particles suspended in water for 5 days (Fig. 1) showed that the majority of particles underwent ligand exchange (Table 1, entry 8). ATOFMS analysis of these samples showed that >50% of the MOF particles contained both Br and CN ion fragments (Fig. 2, Fig. S3†), indicating that more than half of the UiO-66 particles had undergone ligand exchange. At an elevated temperature (85 °C) in water nearly all of the particles underwent ligand exchange (Table 1, entry 10).|
|
|||
|---|---|---|---|
| Entry | Conditionsa | Number of particles—exchanged![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :UiO-66-Br:UiO-66-NH2 :UiO-66-Br:UiO-66-NH2 |
Percentage of particles exchanged (±2%)b |
| a UiO-66-NH2 (0.1 mmol) and UiO-66-Br (0.1 mmol) were placed in a dram vial with 2 mL of the solvent indicated for 5 days. b (number of particles with both ligands)/(total number of particles). | |||
| 1 | CHCl3, RT | 387:548:898 |
21% |
| 2 | CHCl3, 55 °C | 785:469:583 |
43% |
| 3 | MeOH, RT | 677:644:540 |
36% |
| 4 | MeOH, 55 °C | 1243:250:486 |
63% |
| 5 | DMF, RT | 1165:235:606 |
58% |
| 6 | DMF, 55 °C | 1344:259:336 |
69% |
| 7 | DMF, 85 °C | 1419:187:530 |
66% |
| 8 | H2O, RT | 882:420:332 |
54% |
| 9 | H2O, 55 °C | 1549:119:276 |
80% |
| 10 | H2O, 85 °C | 1907:13:56 |
97% |
Complete characterization of the exchanged UiO-66-(Br)(NH2) particles (Table 1, entry 10) confirmed that the structural stability of the material was not compromised by the ligand exchange process. PXRD showed that the crystallinity of exchanged UiO-66-(Br)(NH2) is retained (Fig. 3) and TGA analysis indicates that the material has a similar thermal stability to other UiO-66 MOFs (Fig. S4†). These results indicate that even ‘inert’ MOFs can undergo ligand exchange while maintaining bulk crystallinity. This detail could only be uncovered by a single-particle analysis method (i.e. ATOFMS) and is not revealed by bulk analysis methods such as PXRD. Although ATOFMS was used to unambiguously determine whether a particle contains NH2-BDC, Br-BDC, or both ligands, it is important to note that the ATOFMS method is unable to quantify the ratio of Br-BDC/NH2-BDC in any given particle. Because of fluctuations in laser intensity27 and matrix effects,28 the ratio of Br-BDC/NH2-BDC ion signal intensities cannot be accurately determined. Therefore, ATOFMS was used to determine the number of particles that underwent ligand exchange (e.g. >50% in water for 5 days), but not the fraction of ligands exchanged per particle. However, our experiments utilizing postsynthetic ligand exchange (see below) indicate that a substantial fraction of the ligands in a single particle can be replaced via this process.
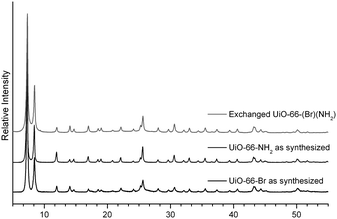 | ||
| Fig. 3 PXRD patterns of UiO-66-Br, UiO-66-NH2, and exchanged UiO-66-(Br)(NH2). The exchanged UiO-66-(Br)(NH2) sample was produced in water at 85 °C after 5 days. | ||
Chloroform, methanol, N,N-dimethylformamide (DMF), and water were used to examine the effect of the solvent on particle-to-particle ligand exchange. These solvents were selected because they are often used for the synthesis, activation, and postsynthetic modification (PSM) of MOFs.22Spectra from ∼2000 single particles were grouped into clusters using a hierarchical clustering algorithm; clusters were manually classified into one of the three categories (see the Supporting Information for details†).29 Error analysis was solved assuming Poisson statistics. The exchange ratio strongly correlated with solvent polarity, with water having the highest exchange ratio followed by DMF, methanol, and finally chloroform having the lowest exchange ratio (Table 1).
The size distribution of the particles could also be determined by ATOFMS (dva, Fig. S5†), which shows that the particle size is not changing during the incubation period. The invariant particle size indicates that the mixing of ligands observed in these experiments is due to ligand exchange and not particle growth. Overall, the ATOFMS experiments provide new insight into the stability in this important class of purportedly inert materials. The robust UiO-66 MOFs are structurally stable, but can be labile with respect to ligand exchange/composition, in the presence of polar solvents such as water. This observation has significant repercussions in the context of some of the proposed industrial applications of MOFs, where chronic moisture exposure is likely.
Postsynthetic ligand exchange from the liquid phase
To further explore and exploit the observed exchange process, we performed biphasic (solid-liquid) ligand exchange experiments. UiO-66-Br was suspended in a solution of NH2-BDC in water and incubated for 5 days at several different temperatures. The NH2-BDC solution and solid UiO-66-Br material were then separated and analyzed by 1H NMR (the UiO-66-Br solid was dissolved with HF before analysis, see Supporting Information†). Consistent with our previous findings, both Br-BDC and NH2-BDC were found in the aqueous solution and in the solid UiO-66 MOF (Table 2). The extent of replacement of NH2-BDC for Br-BDC in the UiO-66 material was temperature dependent. For example, at room temperature the amount of NH2-BDC incorporated into UiO-66-Br (∼9%) was identical to the amount of Br-BDC found leached into to liquid phase (∼9%), consistent with a ligand exchange process (Table 2). It is important to note that the NH2-BDC ligand had to be administered in a deprotonated form in order to observe ligand exchange (see Supporting Information†). At 55 °C and 85 °C, 63% and 76% of the Br-BDC ligands, respectively, in the original framework had been replaced by NH2-BDC as determined by 1H NMR (Table 2). Interestingly, exchange of Br-BDC into UiO-66-NH2 proceeded to a somewhat lesser degree (43% at 85 °C, Fig. S6†). In the light of this observation, experiments were performed comparing exchange of either NH2-BDC or Br-BDC with unfunctionalized UiO-66 (Fig. S7†). Consistent with the observations above, NH2-BDC showed a higher degree of exchange than Br-BDC. Br-BDC was found to be ∼48% incorporated into UiO-66, while NH2-BDC was ∼67% exchanged into the material (r.t., 5 days). The difference in exchange ability is likely due to a combination of factors including differences in donor ability, sterics, and solubility of the NH2-BDCversusBr-BDC ligand. Overall, these observations clearly show that PSE has a remarkable capacity to extensively modify the MOF, demonstrating far more than just superficial (i.e. surface) changes to the crystalline material. These results demonstrate that we can obtain mixed, bifunctional MOFs viaPSE, using temperature and time to modulate the degree of ligand replacement.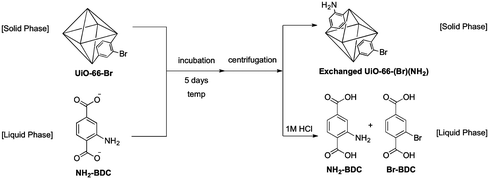
Several observations and experiments confirmed that this PSE truly involves ligand exchange and not a particle growth process. First, in these experiments only the ligand was added (NH2-BDC), but no additional Zr(IV) source that would permit particle growth. Second, both scanning electron microscopy (SEM) and dynamic light scattering (DLS) experiments confirmed that the particle size remains unchanged during ligand exchange, as no significant particle size increase or decrease was observed via either technique (Fig. S8 and S9†). Lastly, we found that UiO-66 frameworks could not be synthesized de novo under the conditions employed for the exchange reaction (r.t., 55 °C, 85 °C) with BDC ligands and Zr(IV) sources. Even the seeding of these reactions with UiO-66 (∼1 mg) did not produce framework crystallites under the exchange reaction conditions. All of these experiments support the idea that PSE is occurring in a SCSC (or single particle-to-particle) fashion and not through a framework growth or dissolution-reassembly mechanism.
Postsynthetic exchange functionalization
Having demonstrated that these MOFs could undergo ligand exchange in a phase separated (solid-liquid) fashion, we explored PSE as an approach to introduce useful functional groups into the MOF (Fig. 4). At first, we focused our efforts on azide-substituted ligands, which are thermally unstable and could be dangerous to use, under standard solvothermal conditions. Azides are attractive groups to include in MOFs because of their use in ‘click’ chemistry30–32 and their photoreactivity.33 A recent report describes the introduction of azides into amine-bearing MOFs in a PSM fashion.34 To utilize PSE, UiO-66 was exposed to an aqueous solution of 2-azido-1,4-benzenedicarboxylate (N3-BDC) under ambient conditions (Fig. 4). After separating the solids by centrifugation and washing with methanol, the presence of the azide moiety in the MOF particles was confirmed by 1H NMR, ESI-MS, and FT-IR (Fig. S10–S12†). The degree of PSE was time dependent: ∼9% after 1 day, ∼22% after 2 days, and ∼50% after 5 days, respectively (Fig. S13†). The crystallinity of UiO-66 was completely retained after exchange, as confirmed by PXRD and BET surface area values (Fig. S14 and Table S1†).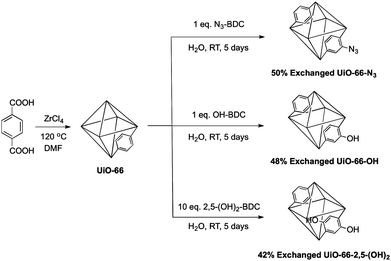
To further explore the scope of PSE we employed several hydroxyl BDC ligands in these reactions. Due to the metal binding affinity of the phenol group, MOFs containing free hydroxyl groups are rather uncommon.35UiO-66 was treated with one equivalent of either 2-hydroxy-1,4-benzendicarboxylate (OH-BDC) or 2,5-dihydroxy-1,4-benzenedicarboxylate (2,5-(OH)2-BDC) under the aforementioned PSE conditions. 1H NMR and ESI-MS (Fig. 4, S15, S16†) showed that 48 ± 3% of OH-BDC was incorporated into the UiO-66 framework. The 2,5-(OH)2-BDC ligand gave a much lower degree of incorporation (∼15%) than either N3-BDC or OH-BDC. However, increasing the amount of 2,5-(OH)2-BDC to 10 equivalents gave a material with 42 ± 1% incorporation of 2,5-(OH)2-BDC into the UiO-66 framework. The crystallinity and porosity of 48% exchanged UiO-66-OH and 42% exchanged UiO-66-2,5-(OH)2 were confirmed by PXRD and BET surface area measurements (Fig. S14 and Table S1†). Importantly, UiO-66-OH and UiO-66-2,5-(OH)2 could not be synthesized directly under standard solvothermal conditions in DMF from OH-BDC and 2,5-(OH)2-BDC (Fig. S17†). Using N,N-diethylformamide (DEF) as a solvent at 120 °C, we could obtain solid materials for both OH-BDC and 2,5-(OH)2-BDC; however, the material prepared directly by solvothermal synthesis was of much poorer crystallinity than that obtained via the PSE route (Fig. S14 and S17†). Overall, this study demonstrates that ligand exchange, under ambient conditions, even with relatively inert MOFs, represents a viable strategy for modification of these materials. These findings show that PSE is a new route to obtain functional MOFs, which will serve to compliment other recently reported approaches to mixed, multi-functional MOFs.26,36,37 Although the degree of functionalization achieved by PSE (i.e. percent conversion) is perhaps less than that reported by PSM methods, the experiments reported here show PSE can introduce functional groups not readily obtained by PSM. PSM is generally limited to the organic transformations that can be performed on the functional group ‘tags’ present in the parent MOF (e.g., amine to amide, halo to cyano, etc.). In contrast, PSE is not bound by this limitation, that is, in principle any functionalized ligand can be directly introduced into an existing framework. Furthermore, our findings show the usefulness of PSE with even inert metal-carboxylate bonds; ongoing studies will be required to determine the overall applicability of PSE to different MOFs.
Conclusions
We have developed a new, postsynthetic functionalization method for MOFs under ambient conditions by using ligand exchange. The ligand exchange phenomenon between intact UiO-66 microcrystals was first detected by using ATOFMS, a powerful tool for examining the composition of MOF particles at the single-particle level. The ability to interrogate individual particles uncovered that even highly robust MOFs, like the UiO-66 series, are dynamic under certain conditions. Although the bulk material remains crystalline, in polar solvents these materials readily undergo ligand exchange. Building on these observations, we were able to show that PSE could be used to directly functionalize MOFs under mild reaction conditions. PSE was effective for introducing azide- and hydroxyl-functionalized ligand into UiO-66, which could not be readily prepared with similar quality by direct solvothermal methods. The quantification of ligand exchange was determined and showed that ligand exchange was substantial, with exchange yields of >40% being routinely achieved. Several mechanistic clues indicate that this process does occur by ligand exchange, and not a crystal growth process. The discovery of extensive ligand exchange in the inert MOF UiO-66 opens up a new synthetic approach for multi-functional MOFs, as well as providing important insight into the stability of MOFs, showing that the considerations of crystallinity and compositional stability must be addressed separately.26Acknowledgements
We thank the U.C. San Diego mass facility for assisting with the mass spectrometry experiments. This work was supported by a grant from the Department of Energy (BES Grant No. DE-FG02-08ER46519).Notes and references
- L. J. Murray, M. Dinca and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC
.
- J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC
- L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248–1256 RSC
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef
- G. Ferey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surble and I. Margiolaki, Science, 2005, 309, 2040–2042 CrossRef CAS
- R. Banerjee, A. Phan, B. Wang, C. Knobler, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Science, 2008, 319, 939–943 CrossRef CAS
- A. Schaate, P. Roy, A. Godt, J. Lippke, F. Waltz, M. Wiebcke and P. Behrens, Chem.–Eur. J., 2011, 17, 6643–6651 CrossRef CAS
- E. Gard, J. E. Mayer, B. D. Morrical, T. Dienes, D. P. Fergenson and K. A. Prather, Anal. Chem., 1997, 69, 4083–4091 CrossRef CAS
- H. J. Park, Y. E. Cheon and M. P. Suh, Chem.–Eur. J., 2010, 16, 11662–11669 CrossRef CAS
- M. Kondo, S. Furukawa, K. Hirai and S. Kitagawa, Angew. Chem., Int. Ed., 2010, 49, 5327–5330 CrossRef CAS
- J. Seo, C. Bonneau, R. Matsuda, M. Takata and S. Kitagawa, J. Am. Chem. Soc., 2011, 133, 9005–9013 CrossRef CAS
- S. Das, H. Kim and K. Kim, J. Am. Chem. Soc., 2009, 131, 3814–3815 CrossRef CAS
- S. Jiang, J. T. A. Jones, T. Hasell, C. E. Blythe, D. J. Adams, A. Trewin and A. I. Cooper, Nat. Commun., 2011, 2, 207 CrossRef
- B. J. Burnett, P. M. Barron, C. Hu and W. Choe, J. Am. Chem. Soc., 2011, 133, 9984–9987 CrossRef CAS
- J. R. Li, D. J. Timmons and H. C. Zhou, J. Am. Chem. Soc., 2009, 131, 6368–6369 CrossRef CAS
- W. Yuan, T. Friscic, D. Apperley and S. L. James, Angew. Chem. Int. Ed., 2010, 49, 3916–3919 CAS
- V. Guillerm, S. Gross, C. Serre, T. Devic, M. Bauer and G. Ferey, Chem. Commun., 2010, 46, 767–769 RSC
- P. Horcajada, C. Serre, D. Grosso, C. Boissiere, S. Perruchas, C. Sanchez and G. Ferey, Adv. Mater., 2009, 21, 1931–1935 CrossRef CAS
- J. R. Li and H. C. Zhou, Nat. Chem., 2010, 2, 893–898 CrossRef CAS
- M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, C. Larabi, E. A. Quadrelli, F. Bonino and K. P. Lillerud, Chem. Mater., 2010, 22, 6632–6640 CrossRef CAS
- S. J. Garibay and S. M. Cohen, Chem. Commun., 2010, 46, 7700–7702 RSC
- R. Bahadur, L. M. Russell and K. Prather, Aerosol Sci. Technol., 2010, 44, 551–562 CAS
- Y. Su, M. F. Sipin, K. A. Prather, R. M. Gelein, A. Lunts and G. Oberdorster, Aerosol Sci. Technol., 2005, 39, 400–407 CAS
- M. T. Spencer, H. Furutani, S. J. Oldenburg, T. K. Darlington and K. A. Prather, J. Phys. Chem. C, 2008, 112, 4083–4090 CAS
- M. Kim, J. F. Cahill, K. A. Prather and S. M. Cohen, Chem. Commun., 2011, 47, 7629–7631 RSC
- R. J. Wenzel and K. A. Prather, Rapid Commun. Mass Spectrom., 2004, 18, 1525–1533 CrossRef CAS
- D. S. Gross, M. E. Galli, P. J. Silva and K. A. Prather, Anal. Chem., 2000, 72, 416–422 CrossRef CAS
- D. M. Murphy, A. M. Middlebrook and M. Warshawsky, Aerosol Sci. Technol., 2003, 37, 382–391 CAS
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS
- T. Gadzikwa, O. K. Farha, C. D. Malliakas, M. G. Kanatzidis, J. T. Hupp and S. T. Nguyen, J. Am. Chem. Soc., 2009, 131, 13613–13615 CrossRef CAS
- Y. Goto, H. Sato, S. Shinkai and K. Sada, J. Am. Chem. Soc., 2008, 130, 14354–14355 CrossRef CAS
- H. Sato, R. Matsuda, K. Sugimoto, M. Takata and S. Kitagawa, Nat. Mater., 2010, 9, 661–666 CrossRef CAS
- M. Savonnet, D. Bazer-Bachi, N. Bats, J. Perez-Pellitero, E. Jeanneau, V. Lecocq, C. Pinel and D. Farrusseng, J. Am. Chem. Soc., 2010, 132, 4518–4519 CrossRef CAS
- K. K. Tanabe, C. A. Allen and S. M. Cohen, Angew. Chem., Int. Ed., 2010, 49, 9730–9733 CrossRef CAS
- H. Deng, C. J. Doonan, H. Furukawa, R. B. Ferreira, J. Towne, C. B. Knobler, B. Wang and O. M. Yaghi, Science, 2010, 327, 846–850 CrossRef CAS
- A. D. Burrows, CrystEngComm, 2011, 13, 3623–3642 RSC
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1sc00394a |
| ‡ M. K. and J. F. C. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2012 |