A novel route for self-assembly of gold nanoparticle–TiO2 nanotube array (Au/TNTs) heterostructure for versatile catalytic applications: pinpoint position via hierarchically dendritic ligand†
Fang-Xing
Xiao
*
State Key Laboratory Breeding Base of Photocatalysis, College of Chemistry and Chemical Engineering, Fuzhou University, Fuzhou, 350002, P. R. China. E-mail: fangxing2010@gmail.com
First published on 26th October 2012
Abstract
We have developed an efficient self-assembly strategy to readily achieve well-defined Au/TNTs heterostructure based on robust as well as multilayered dendritic dithiolated diethylenetriaminepentaacetic (DTDTPA) ligand as bridging medium. Versatile catalytic performances of the hybrid system were investigated.
In recent years, one-dimensional (1-D) nanostructures, such as nanowire, nanotube, and nanobelt have garnered tremendous attention owing to their alluring potential applications in diverse fields.1–3 Titania nanotubes (TiO2 NTs), as quintessential 1-D nanomaterials, have been touted as versatile photocatalysts for degradation of pollutants in air or water and for H2 production.4,5 Driven by this, considerable efforts have been devoted to the synthesis of various 1-D TiO2 NTs, in which TiO2 nanotube arrays (TNTs) fabricated via electrochemical anodization appear to be the most promising candidates for photocatalytic applications or solar energy conversion.6–8
While transformation of TiO2 morphology to nanotubular structure may contribute to enhanced photocatalytic efficiency compared to commonly used TiO2 nanoparticles (NPs), it is far more difficult to fundamentally tackle the core issue of photocatalysis.9 Specifically, the significant breakthrough in suppressing the recombination of photogenerated electron-hole pairs remains the key challenge. To this end, research activities have been focused on improving the photocatalytic activity of TNTs, in which modifications of TNTs with noble metal NPs have been demonstrated to be an elegant way to circumvent this issue.10–13 Unfortunately, in most cases, including electrodeposition,14,15 dipping-calcination,16,17 photoreduction18 and deposition-precipitation,19 these synthetic protocols are normally time-consuming and complicated. Additionally, it is well-established that monodispersed deposition of metal NPs on the framework of TNTs has so far remained unsatisfactory, and, especially, uniform deposition of metal NPs on the interior surface of TNTs is evidenced to be unfavorable because of poor entry to the TNTs.20 Furthermore, deposited metal NPs generally suffer from serious agglomeration along with uncontrolled size and uneven distribution upon post-calcination, thereby rendering preparation of well-defined metal/TNTs heterostructures difficult. It is thus highly desirable to develop an efficient approach to achieve the construction of hierarchical metal/TNTs heterostructures. On the other hand, tailor-made Au NPs passivated with dendritic ligands have been extensively investigated showing fascinating physico-chemical properties.21–23 Nonetheless, how to fully harness the capped ligands to induce spontaneous deposition of Au NPs on 1-D semiconductoring nanomaterials via facile self-assembly engineering has not yet been reported.24
Herein, we demonstrate a facile route for self-assembly of Au NPs on the TNTs which were prepared by electrochemical anodization, based on pronounced electrostatic force afforded by positively charged TNTs and negatively charged diethylenetriaminepentaacetic (DTDTPA) profile, leading to a well-defined Au/TNTs heterostructure. The monodispersity of Au NPs can be retained after removing the surface ligand via post-heat treatment, and the thus-obtained heterostructure exhibits versatile catalytic performances under ambient conditions.
Fig. 1 depicts the fabrication process of the Au/TNTs heterostructure via a self-assembly approach. The synthesis and characterizations of Au@DTDTPA, TNTs, and Au/TNTs are elucidated in the ESI† (Experimental section). The DTDTPA capped on the Au NP (ca. 2.8 nm, Fig. S1†) is mainly composed of a large amount of carboxyl groups which can be completely deprotonated under optimal pH conditions, resulting in hierarchically-dendritic negatively-charged profile (Fig. S2†). Au@DTDTPA demonstrates excellent stability and pronounced negative zeta potential value (<−30 mV) in the pH realm of 6–10.24 With respect to TNTs, the positively charged layer can be readily achieved by immersing TNTs in an acid medium with pH lower than the isoelectric point (IEP) of TiO2 (4.5–6.8).25 It is thus expected that the self-assembly process induced by electrostatic force would be initiated between the negatively charged Au@DTDTPA and positively charged TNTs. However, no Au NPs were observed to self-assemble to the TNTs under the current experimental conditions (Fig. S3†, pH = 6), which is mainly ascribed to incompatibility between IEP of TNTs and pH of Au@DTDTPA (i.e. 4.5–6.8 vs. 6–10). As a result, it is essential to reinforce the positive charge intensity of the TiO2 layer in TNTs to surmount the limitation, then the self-assembly process could be spontaneously initiated. In this regard, UV light irradiation was harnessed to increase surface hydroxyl groups (–OH) of TNTs, since it has been evidenced that hydroxylation of TiO2 surface can be obtained by exposing surface to the UV light (λ < 382 nm);26,27 thus sufficient hydrophilic conversion of TNTs surface was realized (Fig. S4†).28 Moreover, with a view to achieving a pronounced positively charged surface terminated with –NH3+ groups (Fig. S4†), the UV-treated TNTs were further modified with 3-aminopropyltrimethoxysilane (APS) via reaction between OH group (from the outer wall surface of TNTs) and CH3O–Si– (from APS). Meanwhile, a large amount of carboxyl groups completely deprotonated in the DTDTPA renders Au@DTDTPA a potential negatively charged candidate for pinpoint deposition to the APS-UV-treated TNTs. Therefore, a spontaneous self-assembly scenario, based on substantial electrostatic force afforded by opposite charged constituents, chemical bonding (N–Au) and hydrogen bonding (COOH from DTDTPA and OH from TiO2), allows for rapid deposition of Au@DTDTPA on the TNTs (Fig. 1). More significantly, the robust multilayered DTDTPA ligand endows Au NPs with anti-agglomeration properties, retaining monodispersity of Au NPs on the framework of TNTs after calcination.
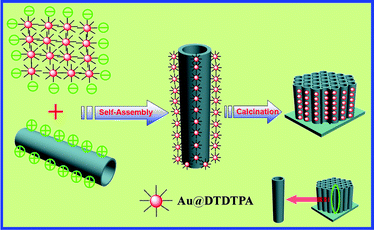 | ||
| Fig. 1 Schematic illustration showing the self-assembly of the hierarchical Au/TNTs heterostructure. | ||
As shown in Fig. 2a–2c, pristine TNTs exhibit a vertically oriented nanotubular structure with thickness profile of ca. 6 μm and mean pore diameter of ca. 36 nm growing vertically from the Ti foil. The HRTEM images in Fig. 2f–2g reveal lattice fringes of 3.52 Å and 2.33 Å, which are attributed the to (101) and (111) crystallographic planes of anatase TiO2 and the face-centered cubic (fcc) structure of Au, respectively. The HRTEM image of single Au NP was separately provided in Fig. S5†. The EDX patterns in Fig. S6† exhibit signals of Ti and O from TiO2 and Au from Au NPs. It is noteworthy that Au NPs self-assembling to the framework of TNTs, especially when the surface profile was completely removed by calcination, retain excellent monodispersity (Fig. 2d–2f), leading to a well-defined Au/TNTs heterostructure. The well-defined morphology of the Au/TNTs heterostructure is speculated to principally arise from intimate interaction between Au@DTDTPA and TNTs which can be unveiled by XPS (Fig. S7†). As shown in Table S1†, conspicuous binding energy shifts of 0.36 eV (531.40 eV vs. 531.04 eV) and 0.53 eV (532.30 eV vs. 531.83 eV) in terms of Ti–OH and C–OH groups in TNTs were clearly observed after deposition of Au@DTDTPA, indicating intimate interfacial interaction between TNTs and DTDTPA based on pronounced electrostatic force and hydrogen bonding.24 Moreover, in order to highlight the contribution of DTDTPA as a target-directing and anti-agglomeration medium during the self-assembly process, citrate-capped Au NPs (Au@Citrate) with a less carboxylic acid-derivatized surface were also used for comparison. Notably, sparse Au NPs with larger diameter (ca. 25 nm) were tethered to the TNTs after the same heat-treatment (Fig. S8†), which is predominantly due to much a weaker electrostatic force afforded by citrate species compared with DTDTPA, thereby resulting in fewer Au NPs anchored on the TNTs. Hence, the significant role of DTDTPA was substantiated.
 | ||
| Fig. 2 (a) Panoramic SEM and (b–c) TEM views of blank TNTs with cross-sectional image in the inset of (a), (d) SEM and (e–g) TEM views of the Au/TNTs heterostructure. | ||
UV-vis diffuse reflectance spectra (DRS) of TNTs and Au/TNTs heterostructure (Fig. S9†) confirm successful self-assembly of Au NPs on the TiO2 scaffold and substantial photosensitivity in the UV-light range. The FTIR of the Au@DTDTPA/TNTs nanostructure also corroborates the intimate attachment of Au@DTDTPA on the TNTs (Fig. S10†). XRD patterns of TNTs and Au/TNTs heterostructure (Fig. S11†) reveal that all the diffraction peaks can be indexed to anatase TiO2 (JCPDS NO. 21-1272) and Ti phase (JCPDS NO. 44-1294) for TNTs and fcc-centered cubic phase of Au (JCPDS NO. 65-2870), demonstrating faithful agreement with high-resolution XPS spectra of Ti 2p (Fig. S7–f†) and Au 4f (Fig. S7–g†) of the Au@DTDTPA/TNTs, which discloses the textural properties of anatase TiO2 and metallic Au. In this respect, it is expected that the well-defined Au/TNTs heterostructure will exhibit appealing catalytic activities in view of its good monodispersed deposition of Au NPs on the TNTs.
Catalytic reduction of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP) under ambient conditions was used as a model reaction for evaluation of catalytic activities of the metal NP-deposited nanocomposites, which follows pseudo-first-order kinetics.29Fig. 3a shows that the catalytic reduction activities of Au/TNTs and Au@DTDTPA/TNTs are remarkably higher than that of TNTs, resulting from active Au NPs uniformly anchored on the TNTs. It should be noted that more enhanced catalytic activity is attained when the ligand was removed from the Au@DTDTPA/TNTs heterostructure, which is attributed to exposure of more highly active Au facets. Fig. 3b reveals the photodegradation of methyl orange (MO) over the samples under UV light irradiation (365 ± 15 nm), in which kinetic rate constant follows the order Au/TNTs > Au@DTDTPA/TNTs ≈ TNTs. It is reasonably speculated that monodispersed Au NPs may play imperative roles as “electron reservoirs” to reinforce the separation of photoexcited electron-hole carriers, giving rise to markedly enhanced photocatalytic activity of the Au/TNTs heterostructure, which can be further verified by the photoelectrochemical results.
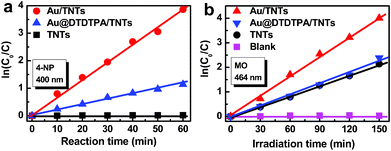 | ||
| Fig. 3 (a) Catalytic reduction of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP) over TNTs, Au@DTDTPA/TNTs and Au/TNTs heterostructure with an excess amount of NaBH4 in aqueous media at ambient temperature by monitoring the decrease in the absorption intensity of 4-NP for the peak at 400 nm. (b) Photocatalytic performances of TNTs, Au@DTDTPA/TNTs and Au/TNTs heterostructure under the irradiation of UV light (365 ± 15 nm). | ||
Fig. 4a shows that the transient photocurrent response of Au/TNTs heterostructure electrode in several on-off cycles of UV light irradiation is significantly reinforced in comparison with blank TNTs, and, moreover, impedance arc radius of Au/TNTs heterostructure (Fig. 4b) is smaller than that for TNTs. It is worthwhile to note that the arc radii order of the electrodes agrees well with the order for photocatalytic activities of the samples. In this regard, both photocurrent and electrochemical impedance spectroscopy (EIS) results indicate that remarkably enhanced photocatalytic performance of the Au/TNTs heterostructure can be ascribed to the role of Au NPs acting as “electron trap”, thus prolonging separation lifetime of photoexcited electron-hole charge carriers.
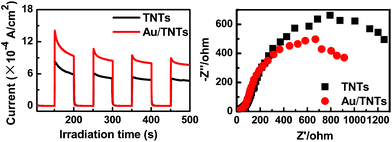 | ||
| Fig. 4 (a) Transient photocurrent response of TNTs and Au/TNTs heterostructure in 0.1 M Na2SO4 aqueous solution under UV light irradiation (365 ± 15 nm). The potential of the working electrode was set at 0.0 V versus the Pt counter electrode. (b) Electrochemical impedance spectroscopy (EIS) Nynquist plots of TNTs and Au/TNTs heterostructure electrodes under UV light illumination (365 ± 15 nm). The amplitude of the sinusoidal wave was set at 10 mV and the frequency varied from 100 kHz to 0.05 Hz. | ||
In summary, well-defined Au/TNTs heterostructures have been prepared via elegant self-assembly of Au@DTDTPA to the framework of TNTs based on pronounced electrostatic force, in which negatively charged multilayered DTDTPA plays the vital role for spontaneous monodispersed deposition and retention of Au NPs on the TNTs. Moreover, this hybrid self-assembled system exhibits versatile catalytic performances under ambient conditions. It is hoped that this novel self-assembly route may open up a new avenue for fabrication of metal NP/1-D semiconducting material nanostructures via surface ligand engineering.
Acknowledgements
Financial support by the Program National Basic Research Program of China (973 Program: 2007CB613306) is greatly acknowledged.References
- Y. N. Xia, P. D. Yang, Y. G. Sun, Y. Y. Wu, B. Mayers, B. Gates, Y. D. Yin, F. Kim and H. Q. Yan, Adv. Mater., 2003, 15, 353 CrossRef CAS.
- P. Yang, M. Fardy and R. Yan, Nano Lett., 2010, 10, 1529 CrossRef CAS.
- Z. R. Tang, Y. H. Zhang and Y. J. Xu, ACS Appl. Mater. Interfaces, 2012, 4, 1512 CrossRef CAS.
- M. R. Hoffmann, S. T. Marin, W. Choi and D. W. Bahnemannt, Chem. Rev., 1995, 95, 69 CrossRef CAS.
- M. A. Fox and M. T. Dulay, Chem. Rev., 1993, 93, 341 CrossRef CAS.
- D. Gong, C. A. Grimes, O. K. Varghese, W. C. Hu, R. S. Singh, Z. Chen and E. C. Dickey, J. Mater. Res., 2001, 16, 3331 CrossRef CAS.
- K. Zhu, T. B. Vinzant, N. R. Neale and A. J. Frank, Nano Lett., 2007, 7, 3739 CrossRef CAS.
- A. E. R. Mohamed and S. Rohani, Energy Environ. Sci., 2011, 4, 1065 RSC.
- F. X. Xiao, J. Mater. Chem., 2012, 22, 7819 RSC.
- I. Paramasivam, J. M. Macak and P. Schmuki, Electrochem. Commun., 2008, 10, 71 CrossRef CAS.
- J. Yu, J. Xiong, B. Cheng and S. Liu, Appl. Catal., B, 2005, 60, 211 CrossRef.
- V. Subramanian, E. Wolf and P. V. Kamat, J. Phys. Chem. B, 2001, 105, 11439 CrossRef CAS.
- L. Yang, D. He, Q. Cai and C. A. Grimes, J. Phys. Chem. C, 2007, 111, 8214 CrossRef CAS.
- Y. K. Lai, H. F. Zhuang, K. P. Xie, D. G. Gong, Y. X. Tang, L. Sun, C. J. Lin and Z. Chen, New J. Chem., 2010, 34, 1335 RSC.
- T. G. S. Babu, P. V. Suneesh, T. Ramachandran and B. Nair, Anal. Lett., 2010, 43, 2809 CrossRef CAS.
- Z. H. Xu, J. G. Yu and G. Liu, Electrochem. Commun., 2011, 13, 1260 CrossRef CAS.
- J. H. Lee, H. S. Choi, J. H. Lee, Y. J. Kim, S. J. Suh, C. S. Chi and H. J. Oh, J. Cryst. Growth, 2009, 311, 638 CrossRef CAS.
- L. Sun, J. Li, C. Wang, S. Li, Y. Lai, H. Chen and C. Lin, J. Hazard. Mater., 2009, 171, 1045 CrossRef CAS.
- M. A. Elmoula, E. Panaitescu, M. Phan, D. Yin, C. Richter, L. H. Lewis and L. Menon, J. Mater. Chem., 2009, 19, 4483 RSC.
- J. A. Seabold, K. Shankar, R. H. T. Wilke, M. Paulose, O. K. Varghese and C. A. Grimes, Chem. Mater., 2008, 20, 5266 CrossRef CAS.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293 CrossRef CAS.
- E. Boisselier and D. Astruc, Chem. Soc. Rev., 2009, 38, 1759 RSC.
- L. Moriggi, C. Cannizzo, E. Dumas, C. R. Mayer, A. Ulianov and L. Helm, J. Am. Chem. Soc., 2009, 131, 10828 CrossRef CAS.
- F. X. Xiao, F. C. Wang, X. Z. Fu and Y. Zheng, J. Mater. Chem., 2012, 22, 7819 RSC.
- J. He, R. Mosurkal, L. A. Samuelson, L. Li and J. Kumar, Langmuir, 2003, 19, 2169 CrossRef CAS.
- R. Wang, K. Hashimoto, A. Fujishima, M. Chikuni, E. Kojima, A. Kitamura, M. Shimohigoshi and T. Watanabe, Nature, 1997, 388, 431 CrossRef CAS.
- R. Wang, K. Hashimoto, A. Fujishima, M. Chikuni, E. Kojima, A. Kitamura, M. Shimohigoshi and T. Watanabe, Adv. Mater., 1998, 10, 135 CrossRef CAS.
- J. Zuo and E. Torres, Langmuir, 2010, 26, 15161 Search PubMed.
- T. Yu, J. Zeng, B. Lim and Y. N. Xia, Adv. Mater., 2010, 22, 5188 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details for synthesis of Au@DTDTPA, TNTs, characterization results for the Au/TNTs heterostructure using UV-vis, XRD, XPS, and FTIR. See DOI: 10.1039/c2ra22621a |
| This journal is © The Royal Society of Chemistry 2012 |