DOI:
10.1039/C2RA22616B
(Paper)
RSC Adv., 2012,
2, 12892-12898
One step thermal conversion of lignin to the gasoline range liquid products by using zeolites as additives†
Received
24th October 2012
, Accepted 24th October 2012
First published on 6th November 2012
Abstract
One step thermal conversion of lignin to gasoline range liquid products was accomplished by pyrolyzing softwood (SW) kraft lignin with select zeolites at 600 °C. Gel Permeation Chromatography (GPC) and Nuclear Magnetic Resonance (NMR) including quantitative 13C, 31P-NMR and Heteronuclear Single-Quantum Correlation (HSQC)-NMR were used to characterize various pyrolysis oils. By employing a zeolite catalyst aliphatic hydroxyl groups decreased by 70–100% in the resulting bio-oil and the content of carboxylic acid groups also decreased by 44–85% in comparison to a control pyrolysis oil generated with no additive. Of the additives studied MFI and MOR zeolites provided the best in situ decarboxylation of bio-oils. The results of 13C-NMR indicated after the use of FAU and BEA zeolites, the pyrolysis oils contained ∼80% less methoxy groups than the native pyrolysis oil, and almost all the oxygen (up to ∼87%) belonged to phenolic hydroxyl groups. In addition, the average molecular weight of these two upgraded pyrolysis oils decreased by ∼60% with respect to the control pyrolysis oil and they had a molecular weight profile in the gasoline range (80–120 g mol−1). By adding MFI, FAU and BEA zeolites, the pyrolysis oils contained some polyaromatic hydrocarbons (PAH). In contrast, there were very limited amount of PAH in FER and MOR upgraded pyrolysis oils and almost no PAH in the native pyrolysis oil.
1. Introduction
With declining petroleum resources, increasing fuel demands and growing concerns about the effects of carbon dioxide emissions from fossil fuels, it is imperative to develop sustainable production of fuels and chemicals. Biomass is a renewable resource for the sustainable production of fuels and chemicals that, to date, have been made primarily from fossil resources. The U.S. Department of Agriculture and U.S. Department of Energy established a vision to derive 25% of chemicals and materials and 20% of transportation fuels from biomass by 2030.1 Lignin is a natural aromatic polymer and a main constituent of lignocellulosics biomass; however, it has received much less biorefining efforts than plant polysaccharides2–4 due to its complexity, biological recalcitrance and its relative thermal stability, as its weight loss occurs in a very wide temperature range from 160–900 °C.5 Furthermore, it is well known that the pyrolysis of lignin yields more char and the least amount of bio-oil when compared to cellulose or whole biomass.6,7 It has been reported that most of the water insoluble and the aromatic C–H groups from the pyrolysis of whole biomass pyrolysis oils can be attributed to the pyrolysis of lignin.6 Therefore, upgrading technologies that convert lignin to a potential replacement for diesel and gasoline could provide an attractive biofuel technology and also provide insight into the conversion of whole biomass to green fuels. One step thermal conversion of lignin to gasoline range (molecular weight is ∼105 g mol−1) liquid products or simple petrochemicals such as benzene, toluene, xylene, phenol and catechol appears to be very pragmatic.
Zeolites are a promising class of additives to improve the properties of pyrolysis oils and it has been examined by several researchers. For example, Murzin's group investigated pyrolysis of pine wood with various zeolites, including Beta (BEA), Y (FAU), ZSM-5 (MFI), Mordenite (MOR) and Ferrierite (FER) zeolites.8–12 They concluded that the mass yield of pyrolysis product was only slightly influenced by the types of zeolite, whereas the chemical compositions of bio-oil were dependent on the structure of the zeolite employed. The content of ketones was higher and the amount of acids and alcohols was lower in the bio-oil when ZSM-5 was used as an additive during the pyrolysis. With using a Mordenite zeolite the content of polyaromatic hydrocarbons was relatively low. Beta, Y and Ferrierite zeolites were more effective at catalyzing dehydration and decarbonylation reactions. Huber's group13–15 also examined the influence of zeolite on the properties of pyrolysis products produced from pine wood, cellulose and glucose. They reported that the thermal conversion of glucose to aromatics was a function of the pore size of the zeolite catalyst. Small pore zeolites did not produce aromatics. In contrast, medium pore size (5.2–5.9 Å) zeolites produce mostly aromatic products. Higher coke, lower aromatics and reduced oxygenated species were observed with the use of large pore zeolites. Zhao et al.16 upgraded the water-insoluble fraction (pyrolytic lignin) from pyrolysis oil of rice husks with several additives, including ZSM-5 and Beta zeolites. They found that compared to the Beta zeolite, ZSM-5 produced more aromatics and less coke. Most of oxygenates were found to be converted to arenes and polycyclic aromatic hydrocarbons with zeolites and this conversion was favored at higher temperatures. French et al.17 used molecular-beam mass spectrometry (MBMS) to analyze the product vapor from pyrolysis of cellulose, straw lignin and ground aspen wood with forty different additives at 400, 500 and 600 °C. They found that the highest yield of hydrocarbons (∼16 wt%) was achieved using metal-substituted ZSM-5 zeolite during pyrolysis and the best-performing catalyst belonged to ZSM-5 zeolite while larger-pore zeolites showed less deoxygenation activity.
The pyrolysis of several biomasses such as, corn stalks,18,19 cassava rhizome,20,21 maple,22,23 poplar,24,25 rice husks,16,26 bamboos,27 empty palm fruit bunch,28 oak,29 beech30,31 and pine 32,33 with various zeolites have also been studied in recent years. Most of the reported additives studies have focused on the pyrolysis of biomass, while only a few have examined the behavior of lignin in the presence of additives during pyrolysis. The goal of this work is to convert lignin to low molecular weight aromatics (molecular weight is ∼100 g mol−1) in one step, which could then be used as a precursor for gasoline and/or the possible substitution of petrochemicals such as phenol and catechol. This was accomplished by using a series of zeolites as pyrolysis additives to influence the chemical components generated during the pyrolysis of lignin. To characterize the whole portions of various pyrolysis oils, we used Nuclear Magnetic Resonance (NMR) including 13C, 31P and Heteronuclear Single-Quantum Correlation (HSQC)-NMR6,34–37 as the analysis technique.
2. Materials and methods
All reagents used in this study were purchased from VWR International or Sigma-Aldrich (St. Louis, MO) and used as received. Lignin was isolated from a commercial USA softwood kraft pulping liquor. Zeolites (CBV 3020E, CP 814, CBV 710, CP 914C and CBV 21A) were purchased from Zeolyst, Inc.
Nowadays, kraft lignin obtained by precipitation from the pulping liquor is abundant and has been used as a low-value material. As the pulp industry modernizes its processes and reduces its overall energy needs, several mills have encountered production limitations due to recovery furnace capacity restraints. To address these and related needs, recent studies have developed a commercial protocol to acid precipitate lignin and recover it as a dry powder and research is ongoing to develop applications for this resource.2–4
Lignin samples used in this study were isolated from a commercial softwood kraft pulping liquor following published methods.34 In brief, the cooking liquor was filtered through filter paper and the filtrate was treated with ethylenediaminetetraacetic acid disodium salt (EDTA-2Na+, 0.50 g/100.00 mL liquor) and stirred for 1 h. The liquor was adjusted to a pH value of 6.0 with 2.0 M H2SO4 and stirred vigorously for 1 h. The liquors were then further acidified to a pH of 3.0 and frozen at −20 °C. After thawing, the precipitates were collected on a medium sintered glass funnel and washed three times with cold water by suspending the precipitates in the water and stirring vigorously at 0 C for 1 h. The precipitates were collected, air dried, and soxhlet extracted with pentane for 24 h. The solid product was air dried and further dried under high vacuum at 45 °C for 48 h. The resulting purified kraft lignin sample was stored at −5 °C.
2.2 Preparation of pyrolysis sample
Different types (MFI, FAU, BEA, FER and MOR) of zeolite were used as the additives in this work. The pyrolysis samples were mechanical stirred lignin with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 weight ratio of zeolites and all the zeolites were activated in the pyrolysis tube at 500 °C under nitrogen for 6 h. Detailed information of each zeolite used can be found in Table 1.
:1 weight ratio of zeolites and all the zeolites were activated in the pyrolysis tube at 500 °C under nitrogen for 6 h. Detailed information of each zeolite used can be found in Table 1.
| |
CBV 3020E |
CBV 720 |
CP 814E |
CP 914C |
CBV 21A |
| SiO2/Al2O3 mole ratio |
30 |
30 |
25 |
20 |
20 |
| Frame work |
MFI |
FAU |
BEA |
FER |
MOR |
| Code name |
Z |
Y |
B |
F |
M |
| Pore dimension |
3 |
3 |
3 |
2 |
1 |
| Pore size (Å) |
5.1 × 5.5 5.3 × 5.6 |
7.4 × 7.4 |
6.6 × 6.7 5.6 × 5.6 |
3.5 × 4.8 4.2 × 5.4 |
6.5 × 7.0 2.6 × 5.7 |
2.3 Equipment and process of pyrolysis
Pyrolysis experiments were conducted in a quartz pyrolysis tube heated with a split-tube furnace. Typically, the pyrolysis sample (4.00 g) was placed in a quartz sample boat that was then positioned in the center of a pyrolysis tube. A K-type thermal couple was immersed in the sample powder during the pyrolysis to measure the heating rate. The pyrolysis tube was flushed with nitrogen gas and the flow rate was adjusted to a value of 500 mL min−1 and then inserted in the pre-heated (600 °C) furnace. The outflow from pyrolysis was passed through two condensers, which were immersed in liquid N2. (Note: liquid N2 was used for experimental convenience) Upon completion of pyrolysis, the reaction tube was removed from the furnace and allowed to cool to room temperature under constant N2 flow. The condensers were then removed from liquid nitrogen. The pyrolysis char and oil were collected for subsequent chemical analysis. In general, the liquid products contained two immiscible phases referred to as heavy and light oil. The light oil was acquired by decantation. The heavy oil is a high molecular weight (∼300 g mol−1) water insoluble organic components mixture and the C:H:O ratio of heavy oil is 10:1:3.35 Due to the high viscosity, it needs to be recovered by washing the reactor with acetone followed by evaporation under reduced pressure. Char yields were determined gravimetrically and gas formation was calculated by mass difference. A summary of the experimental procedure is shown in Fig. 1.
 |
| | Fig. 1 Summary of the experimental procedure. | |
2.4 Characterization of pyrolysis oils by GPC
The weight average molecular weight (Mw), number average molecular weight (Mn) and molecular weight polydispersity of the heavy oils were determined by Gel Permeation Chromatography (GPC) analysis following literature methods.34 Prior to GPC analysis, the heavy oil samples were dissolved in THF (1 mg mL−1) and filtered through a 0.45 μm syringe filter. The samples were injected into a Polymer Standards Service (PSS) Security 1200 system featuring Agilent High-Performance Liquid Chromatography (HPLC) vacuum degasser, isocratic pump, refractive index (RI) detector and UV detector (270 nm). Separation was achieved with four Waters Styragel columns (HR0.5, HR2, HR4, HR6) using tetrahydrofuran (THF) as the mobile phase (1.0 mL min−1) with injection volumes of 30 μL. Data collection and processing were performed using PSS WinGPC Unity software. Molecular weights (Mn and Mw) were calibrated against a calibration curve. The calibration curve was created by fitting a third order polynomial equation to the retention volumes obtained from a series of narrow molecular weight distribution polystyrene standards (i.e., 7.21 × 103, 4.43 × 103, 1.39 × 103, 5.80 × 102 Da), Dioctyl phthalate (Mw = 390 g mol−1), 2,2′-Dihydroxy-4,4′-dimethoxyl-benzophenone (Mw = 274 g mol−1), 2-phenylhydroquinone (Mw = 186 g mol−1), phenol (Mw = 94 g mol−1) and acetone (Mw = 58 g mol−1). The curve fit had an R2 value of 0.998.
2.5 Characterization of pyrolysis oil by NMR
2.5.1 Quantitative 13C-NMR.
All NMR spectral data reported in this study were recorded with a Bruker Avance/DMX 400 MHz NMR spectrometer. Quantitative 13C-NMR were acquired using 100.0 mg heavy oil dissolved in 450 μL methyl sulfoxide-d6 (DMSO-d6) employing an inverse gated decoupling pulse sequence, 90° pulse angle, a pulse delay of 5 s and 6000 scans at room temperature with a line-broadening (LB) of 5.0 Hz. For the pyrolysis oils upgraded by Y and B zeolites, the molecular weight is ∼100 g mol−1 and the longest T1 (T1 was measured using the inversion recovery method and calculated with Bruker's TopSpin software 2.1.) was determined to be 12 s. To reduce the measurement time, 1 mg ml−1 relaxation reagent chromium acetylacetonate was added into the solutions and the longest T1 decreased to 4 s. Therefore, for these two pyrolysis oil samples, the pulse delay was set to be 20 s.
2.5.2 Quantitative 31P-NMR.
Quantitative 31P-NMR were acquired after in situ derivatization of the samples using 10.0 mg of heavy oil or 4.0 mg for the light oil with 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (TMDP) in a solution of (1.6:1 v/v) pyridine/CDCl3, chromium acetylacetonate (relaxation agent) and endo-N-hydroxy-5-norbornene-2,3-dicarboximide (NHND, internal standard). 31P-NMR spectra data was acquired using an inverse gated decoupling pulse sequence, 90° pulse angle, 25 s pulse delay, and 128 scans at room temperature with a LB of 4.0 Hz.
2.5.3 Characterization of pyrolysis oil by HSQC-NMR.
HSQC-NMR were acquired using 100.0 mg pyrolysis oil dissolved in 450 μL DMSO-d6 employing a standard Bruker pulse sequence “hsqcetgpsi.2” with a 90° pulse, 0.11 s acquisition time, a 1.5 s pulse delay, a 1JC–H of 145 Hz, 48 scans and acquisition of 1024 data points (for 1H) and 256 increments (for 13C). The 1H and 13C pulse widths are p1 = 11.30 μs and p3 = 10.00 μs, respectively. The 1H and 13C spectral widths are 13.02 ppm and 220.00 ppm, respectively. The central solvent peak was used for chemical shift calibration. HSQC data processing and plots were carried out using MestReNova v7.1.0 software's default processing template and automatic phase and baseline correction.
3. Results and discussion
3.1 Yields of pyrolysis products
The yields from pyrolyzing a softwood kraft lignin with different zeolites at 600 °C are summarized in Fig. 2 These results indicate that the yields of char increased after the use of additives and pyrolysis of lignin with Y and B zeolites yielded the most char. Pyrolysis with F and M zeolites increased the yield of heavy oil. In contrast, after the use of Z, Y and B zeolites, lignin produced less heavy oil but more light oil. The differences between the yields of pyrolysis products indicate the influence of zeolite on the pyrolysis process is related to the type of zeolite framework.
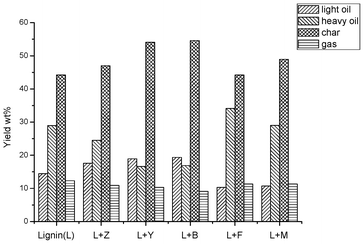 |
| | Fig. 2 Yields (wt%) of light oil, heavy oil, char (excludes the weight of zeolite) and gas for the pyrolysis of pure SW kraft lignin and pyrolysis of SW kraft lignin with 1.0/1.0 (Wadditive/Wlignin) of Z, Y, B, F and M zeolites as additives at 600 °C for 10 min. | |
3.2 Quantitative 31P-NMR analysis of pyrolysis oils
Our previous work34–37 introduced quantitative 31P-NMR to determine hydroxyl functional groups in pyrolysis oils. The 31P-NMR chemical shifts and integration regions of the phosphitylated aryl/alkyl hydroxyl groups and water with 2-chloro-4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaphospholane (TMDP) are summarized in Table 2. The integration results for the heavy oils are summarized in Fig. 3. After the use of Y and B zeolites as additives, the aliphatic hydroxyl groups in the heavy oils were completely decomposed. With the other three types of zeolite, the aliphatic hydroxyl groups also decreased by 70–95%. The results show that for the zeolites studied the resulting pyrolysis oils were significantly dehydrated. Compared to the Z, F and M zeolites, after the use of Y and B zeolites, the heavy oils contained more C5 substituted guaiacyl phenolic, catechol and p-hydroxy-phenyl types of hydroxyl groups but less normal guaiacyl phenolic hydroxyl group. In contrast, with adding F and M zeolites, lignin pyrolysis yielded more normal guaiacyl phenolic but less p-hydroxy-phenyl and C5 substituted guaiacyl phenolic types of hydroxyl groups. Since catechol and p-hydroxy-phenyl types of hydroxyl groups are the decomposition products of methoxyl-aromatic and ether bonds in the lignin structure,34,37 this indicates that the Y and B zeolites could perform the cleavage of aromatic C–O bonds more effectively. After the use of zeolites, the content of carboxylic acid decreased by 44–85%, which is anticipated to be a much more suitable biofuel precursor. Compared to the other studied zeolites, Z and M zeolites could induce decarboxylation reactions more efficiently. Several researchers have reported similarly enhanced dehydration and decarboxylation of model compounds on the surface of zeolite.38–43
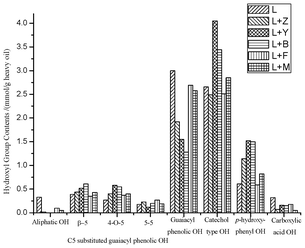 |
| | Fig. 3 Hydroxyl group contents (mmol g−1 of heavy oil) of different heavy oils produced by pyrolysis of pure SW kraft lignin and pyrolysis of SW kraft lignin with 1.0/1.0 (Wadditive/Wlignin) of Z, Y, B, F and M zeolites as additives at 600 °C for 10 min, determined by quantitative 31P-NMR after derivatization with TMDP. | |
Table 2 Chemical shifts and integration regions for lignin pyrolysis oil derivatized with TMDP in a quantitative 31P-NMR34–37
| Functional group |
Integration region (ppm) |
|
endo-N-hydroxy-5-norbornene-2,3-dicarboximide (NHND, internal standard) |
151.0–152.8 |
| Aliphatic OH |
150.0–145.5 |
| C5 Substituted guaiacyl phenolic OH |
β-5 |
144.7–142.8 |
| 4-O-5 |
142.8–141.7 |
| 5-5 |
141.7–140.2 |
| Guaiacyl phenolic OH |
140.2–139.0 |
| Catechol type OH |
139.0–138.2 |
|
p-hydroxy-phenyl OH |
138.2–137.3 |
| Carboxylic acid-OH |
136.6–133.6 |
| Water |
133.1–131.3, 16.9–15.1 |
Our previous studies indicated that the light oils from pyrolysis of lignin contain more than 80 wt% of water and ∼10 wt% of methanol, catechol and acidic acid.34,35,37 The integration results of those four major components in the light oils are summarized in Fig. 4. For all the upgraded light oils, the yields of methanol and acetic acid significantly decreased. In contrast, the content of catechol increased after the use of zeolites. Compare to the Z and F zeolites, the use of Y and B zeolites yielded a light oil with increased catechol which indicates a more efficient decomposition of ether bond and methoxyl group. The content of catechol increased by 320% after the use of Y zeolite, this pure catechol in the light oil could be used as an alternative pathway for its synthesis. As anticipated, the content of the major dehydration product, water increased with the use of zeolites.
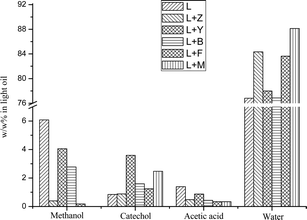 |
| | Fig. 4 Weight percentage of four major components in light oils produced by pyrolysis of pure SW kraft lignin and pyrolysis of SW kraft lignin with 1.0/1.0 (Wadditive/Wlignin) of Z, Y, B, F and M zeolites as additives at 600 °C for 10 min, determined by quantitative 31P-NMR after derivatization with TMDP. | |
3.3 Quantitative 13C-NMR analysis of pyrolysis oils
To fully characterize the functional groups in the heavy oils, a detailed analysis was accomplished by using 13C-NMR. The 13C-NMR chemical shift assignment ranges are based on our previous work34 and shown in the Table 3. The integration results of this analysis for the heavy oils are summarized in Fig. 5. After the use of Y and B zeolites, the heavy oils contained ∼80% less methoxy groups than the native pyrolysis oil, which indicates a very efficient decomposition of methoxyl groups and this result could explain the increasing contents of catechol type and p-hydroxy-phenyl hydroxyl groups detected by 31P-NMR. The two types of methyl aromatic bonds in the heavy oils are reported to be the rearrangement products of methoxyl groups.34 After the use of these two zeolites, the heavy oils contained relatively large amount of methyl aromatic bonds, which is evidence for the enhanced cleavage of methoxyl groups. The 13C-NMR results also show that there were only 13–56% carbonyl groups remaining in the upgraded pyrolysis oils, which is consistent with 31P-NMR results. It has been reported the acidity of pyrolysis oils is mainly (60–70%) derived from carboxylic acids and their content detrimentally impacts the stability of pyrolysis oils.44 The significantly reduced amount of carboxylic acids in the zeolite upgraded pyrolysis oils represents a more stable pyrolysis oil which is expected to cause less corrosion problem.44 With the use of F and M zeolites, the heavy oils contained relatively large amount of aromatic C–O bonds but the least amount of aromatic C–C bonds, which indicate that those two zeolites prefer to cleave aromatic C–C bonds or prevent the formation of such bonds. For the Y and B zeolites upgraded pyrolysis oils, the contents of oxygen substituents including aromatic C–O and aliphatic C–O bonds, carbonyl and methoxyl groups decreased by more than 40%. In addition, almost all the remaining oxygen functionality (up to ∼87%) in these two upgraded pyrolysis oils belongs to the phenolic hydroxyl groups.
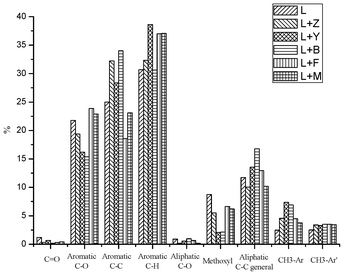 |
| | Fig. 5 Integration results for the SW kraft lignin and the heavy oils produced by pyrolysis of pure SW kraft lignin and pyrolysis of SW kraft lignin with 1.0/1.0 (Wadditive/Wlignin) of Z, Y, B, F and M zeolites as additives at 600 °C for 10 min, detected by quantitative 13C-NMR with using the assignment range showed in Table 3. The results were shown as the percentage of carbon. | |
Table 3
13C-NMR chemical shift assignment range of lignin pyrolysis oil based on the chemical shift database created in our previous work34
| Functional group |
Integration region (ppm) |
| Carbonyl or Carboxyl bond |
215.0–166.5 |
| Aromatic C–O bond |
166.5–142.0 |
| Aromatic C–C bond |
142.0–125.0 |
| Aromatic C–H bond |
125.0–95.8 |
| Aliphatic C–O bond |
95.8–60.8 |
| Methoxyl-Aromatic bond |
60.8–55.2 |
| Aliphatic C–C bond |
General |
55.2–0.0 |
| Methyl – Aromatic (CH3–Ar) |
21.6–19.1 |
| Methyl – Aromatic at ortho position of a hydroxyl or methoxyl group (CH3–Ar′) |
16.1–15.4 |
3.4 HSQC-NMR analysis of pyrolysis oils
To solve spectral overlapping problems when using 13C-NMR to analyze the pyrolysis oils, our previous work demonstrated that HSQC-NMR was uniquely well suited to analyze various C–H bonds present in the pyrolysis oils.6 The HSQC-NMR spectra for the pyrolysis oils after the use of zeolites are shown in Fig. 6–8. HSQC-NMR results show that after the use of Z, Y and B zeolites, the pyrolysis oils contained some polyaromatic hydrocarbons (PAH). In contrast, there were only very limited amount of PAH in the F and M zeolites upgraded pyrolysis oils and almost no PAH in the native pyrolysis oil. The native methoxyl groups (with a hydroxyl group or ether bond in the ortho position) have been completely eliminated after adding Y zeolite during the pyrolysis. The improved degradation of methoxyl groups on the surface of zeolite have been reported in literatures.38,45,46 Since Y type zeolite has a relatively larger pore size and a three dimensional channel system7–9,13,15,22,27,29 which could let small aromatics such as phenol, naphthalene, xylene14,45 go through the channels and thereby improve the upgrading effect more efficiently.46 For the other pyrolysis samples, the decomposition of native type of methoxyl group also has been improved. The content of rearranged methoxyl groups (no hydroxyl group or ether bond in the ortho position) also decreased after the use of Y and B zeolites, which is consisted with the significant reduced amount of methoxyl groups detected by 13C-NMR. Compare to the rearranged methoxyl groups, the zeolites prefer to cleave the native methoxyl groups, which may due to the hydroxyl group in the ortho position facilitates the cleavage. The reported possible degradation pathway of methoxyl groups on the surface of zeolite have been summarized in Fig. 9.38,45,46 After the use of zeolites, there were much more methyl aromatic bonds in the pyrolysis oil which is consistent with our 13C-NMR result. Compare to the native pyrolysis oil, the upgraded pyrolysis oils have relatively lower amount of long chain aliphatic C–C bonds. Zeolite has been reported47–49 to improve the cleavage of aliphatic C–C bonds in the model compounds, which could explain the reduced contents of such bonds in the upgraded pyrolysis oils.
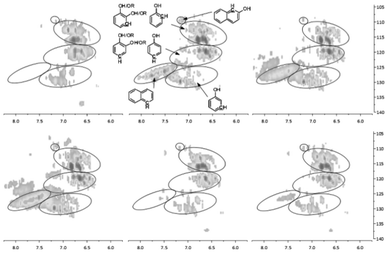 |
| | Fig. 6 Aromatic C–H bonds in the HSQC-NMR spectra for the pyrolysis oils produced by pyrolysis of SW kraft lignin with various zeolites, from left top to right bottom is L, Z, Y, B, F and M upgraded pyrolysis oil. | |
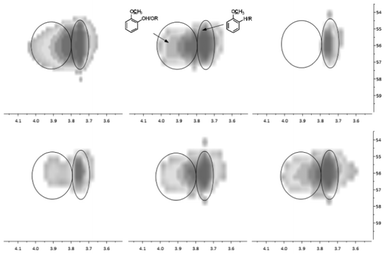 |
| | Fig. 7 Methoxyl groups in the HSQC-NMR spectra for the pyrolysis oils produced by pyrolysis of SW kraft lignin with various zeolites, from left top to right bottom is L, Z, Y, B, F and M upgraded pyrolysis oil. | |
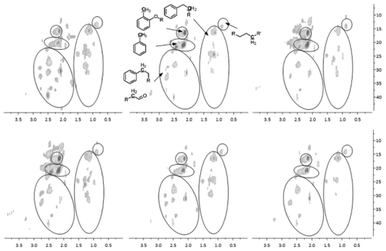 |
| | Fig. 8 Aliphatic C–H bonds in the HSQC-NMR spectra for the pyrolysis oils produced by pyrolysis of SW kraft lignin with various zeolites, from left top to right bottom is L, Z, Y, B, F and M upgraded pyrolysis oil. | |
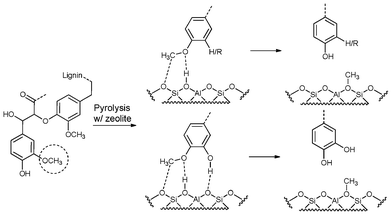 |
| | Fig. 9 The reported possible degradation pathway of methoxyl groups on the surface of zeolite.38,45,46 | |
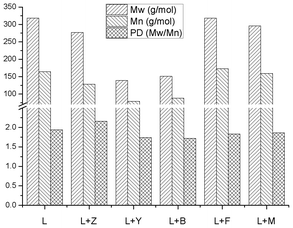 |
| | Fig. 10 Molecular weight distribution and polydispersity of heavy oils produced by pyrolysis of pure SW kraft lignin and pyrolysis of SW kraft lignin with 1.0/1.0 (Wadditive/Wlignin) of Z, Y, B, F and M zeolites as additives at 600 °C for 10 min. | |
4. Conclusion
The pyrolysis of softwood kraft lignin with five different zeolites, including MFI (Z), FAU (Y), BEA (B), FER (F) and MOR (M) as the additives was accomplished at 600 °C. Pyrolysis with F and M zeolites yielded more heavy oil but less light oil. In contrast, after the use of Z, Y and B zeolites, lignin produced less heavy oil but more light oil. The 31P-NMR results show that by using Y and B zeolites, the aliphatic hydroxyl groups in the heavy oils were completely decomposed. With using the other three types of zeolite, the aliphatic hydroxyl groups also decreased by 70–95%. These results indicate zeolites significantly improve the dehydration reactions, which will facilitate the deoxygenation of pyrolysis oil. Compare to the other types of zeolite, after the use of Y and B zeolites, both heavy and light oils contained more catechol type hydroxyl groups, which indicate the Y and B zeolites could perform the cleavage of aromatic C–O bonds more effectively. After using zeolites, the content of carboxylic acid decreased by 44–85%, which represents a much more suitable renewable fuel precursor. Compare to the other types of zeolite, Z and M zeolites prefer to improve the decarboxylation. After the use of Y and B zeolites, the heavy oils contained ∼80% less methoxy groups than the native pyrolysis oil, and almost all the oxygen (up to ∼87%) in these two upgraded pyrolysis oils belongs to the phenolic hydroxyl groups. The HSQC-NMR results show that with adding Z, Y and B zeolites, the pyrolysis oils contained some polyaromatic hydrocarbons (PAH). In contrast, there were very limited amount of PAH in F and M upgraded pyrolysis oils and almost no PAH in the native pyrolysis oil. Compare to rearranged methoxyl groups, all the tested zeolites prefer to cleave the native methoxyl groups, which may due to the hydroxyl groups in the ortho position facilitate the cleavage. The GPC results indicate that the pyrolysis oils upgraded by Y and B zeolites had a molecular weight profile in the gasoline range (80–120 g mol−1).
In summary, the FAU (Y) and BEA (B) zeolites could significantly improve the cleavage of methoxyl-aromatic and ether bonds in the lignin and yield a pyrolysis oil that has a ‘gasoline’ range molecular weight. The upgraded pyrolysis oil could be used as the precursor of gasoline and possible substitution of petrochemicals. The MFI (Z), FER (F) and MOR (M) zeolites could more efficiently decompose the carboxyl groups which will reduce the acidity of pyrolysis oils and make it more suitable for use as a biofuel.
Acknowledgements
The authors would like to acknowledge the financial support from the PSE Fellowship program at IPST@GT and DOE (DE-EE0003144). This work is part of the first author's requirements for the degree of Ph.D. at Georgia Institute of Technology.
References
- R. D. Perlack, L. L. Wright, A. F. Turhollow, R. L. Graham, B. J. Stokes and D. C. Erbach, Biomass as Feedstock for a bioenergy and bioproducts Industry: the technical feasibility of a billion-ton annual supply, 2005 CrossRef.
- M. Nagy, M. Kosa, H. Theliander and A. J. Ragauskas, Green Chem., 2010, 12, 31 RSC.
- P. Tomani, Cellulose Chem. Technol., 2010, 44, 53–58 CAS.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick Jr., J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef CAS.
- H. Yang, R. Yan, H. Chen, D. Lee and C. Zheng, Fuel, 2007, 86, 1781–1788 CrossRef CAS.
- H. Ben and A. J. Ragauskas, Energy Fuels, 2011, 25, 5791–5801 CrossRef CAS.
- S. Czernik and A. V. Bridgwater, Energy Fuels, 2004, 18, 590–598 CrossRef CAS.
- A. Aho, N. Kumar, K. Eränen, T. Salmi, B. Holmbom, P. Backman, M. Hupa and D. Yu Murzin, Biofuels, 2010, 1, 261–273 CrossRef CAS.
- A. Aho, N. Kumar, K. Eranen, T. Salmi, M. Hupa and D. Murzin, Fuel, 2008, 87, 2493–2501 CrossRef CAS.
- A. Aho, N. Kumar, A. V. Lashkul, K. Eränen, M. Ziolek, P. Decyk, T. Salmi, B. Holmbom, M. Hupa and D. Y. Murzin, Fuel, 2010, 89, 1992–2000 CrossRef CAS.
- A. Aho, N. Kumar, K. Eranen, T. Salmi, M. Hupa and D. Murzin, Process Saf. Environ. Prot., 2007, 85, 473–480 CrossRef CAS.
- A. Aho, A. Tokarev, P. Backman, N. Kumar, K. Eränen, M. Hupa, B. Holmbom, T. Salmi and D. Y. Murzin, Top. Catal., 2011, 54, 941–948 CrossRef CAS.
- T. R. Carlson, Y.-T. Cheng, J. Jae and G. W. Huber, Energy Environ. Sci., 2011, 4, 145 CAS.
- J. Jae, G. A. Tompsett, A. J. Foster, K. D. Hammond, S. M. Auerbach, R. F. Lobo and G. W. Huber, J. Catal., 2011, 279, 257–268 CrossRef CAS.
- T. R. Carlson, G. A. Tompsett, W. C. Conner and G. W. Huber, Top. Catal., 2009, 52, 241–252 CrossRef CAS.
- Y. Zhao, L. Deng, B. Liao, Y. Fu and Q.-X. Guo, Energy Fuels, 2010, 24, 5735–5740 CrossRef CAS.
- R. French and S. Czernik, Fuel Process. Technol., 2010, 91, 25–32 CrossRef CAS.
- H. Zhang, R. Xiao, H. Huang and G. Xiao, Bioresour. Technol., 2009, 100, 1428–1434 CrossRef CAS.
- B. B. Uzun and N. Sarioğlu, Fuel Process. Technol., 2009, 90, 705–716 CrossRef CAS.
- A. Pattiya, J. O. Titiloye and A. V. Bridgwater, Fuel, 2010, 89, 244–253 CrossRef CAS.
- A. Pattiya, J. Titiloye and A. Bridgwater, J. Anal. Appl. Pyrolysis, 2008, 81, 72–79 CrossRef CAS.
- J. D. Adjaye and N. N. Bakhshi, Fuel Process. Technol., 1995, 45, 185–202 CrossRef CAS.
- J. D. Adjaye and N. N. Bakhshi, Fuel Process. Technol., 1995, 45, 161–183 CrossRef CAS.
- F. A. Agblevor, S. Beis, O. Mante and N. Abdoulmoumine, Ind. Eng. Chem. Res., 2010, 49, 3533–3538 CrossRef CAS.
- D. Fabbri, A. Adamiano and C. Torri, Anal. Bioanal. Chem., 2010, 397, 309–317 CrossRef CAS.
- P. T. Williams and N. Nugranad, Energy, 2000, 25, 493–513 CrossRef CAS.
- W. Y. Qi, C. W. Hu, G. Y. Li, L. H. Guo, Y. Yang, J. Luo, X. Miao and Y. Du, Green Chem., 2006, 8, 183 RSC.
- M. Misson, R. Haron, M. F. Kamaroddin and N. A. Amin, Bioresour. Technol., 2009, 100, 2867–2873 CrossRef CAS.
- S. Vitolo, M. Seggiani, P. Frediani, G. Ambrosini and L. Politi, Fuel, 1999, 78, 1147–1159 CrossRef CAS.
- E. F. Iliopoulou, E. V. Antonakou, S. A. Karakoulia, I. A. Vasalos, A. A. Lappas and K. S. Triantafyllidis, Chem. Eng. J., 2007, 134, 51–57 CrossRef CAS.
- S. Stephanidis, C. Nitsos, K. Kalogiannis, E. F. Iliopoulou, A. A. Lappas and K. S. Triantafyllidis, Catal. Today, 2011, 167, 37–45 CrossRef CAS.
- B. Valle, A. G. Gayubo, A. s. T. Aguayo, M. Olazar and J. Bilbao, Energy Fuels, 2010, 24, 2060–2070 CrossRef CAS.
- A. Atutxa, R. Aguado, A. G. Gayubo, M. Olazar and J. Bilbao, Energy Fuels, 2005, 19, 765–774 CrossRef CAS.
- H. Ben and A. J. Ragauskas, Energy Fuels, 2011, 25, 2322–2332 CrossRef CAS.
- M. Kosa, H. Ben, H. Theliander and A. J. Ragauskas, Green Chem., 2011, 13, 3196–3202 RSC.
- K. David, H. Ben, J. Muzzy, C. Feik, K. Iisa and A. Ragauskas, Biofuels, 2012, 3, 123–128 CrossRef CAS.
- H. Ben and A. J. Ragauskas, Energy Fuels, 2011, 25, 4662–4668 CrossRef CAS.
- C. A. Mullen, A. A. Boateng, D. J. Mihalcik and N. M. Goldberg, Energy Fuels, 2011, 25, 5444–5451 CrossRef CAS.
- D. J. Mihalcik, A. A. Boateng, C. A. Mullen and N. M. Goldberg, Ind. Eng. Chem. Res., 2011, 50, 13304–13312 CrossRef CAS.
- P. M. Mortensen, J. D. Grunwaldt, P. A. Jensen, K. G. Knudsen and A. D. Jensen, Appl. Catal., A, 2011, 407, 1–19 CrossRef CAS.
- M. A. Peralta, T. Sooknoi, T. Danuthai and D. E. Resasco, J. Mol. Catal. A: Chem., 2009, 312, 78–86 CrossRef CAS.
- H. Chiang and A. Bhan, J. Catal., 2010, 271, 251–261 CrossRef CAS.
- C. Pereira, G. T. Kokotailo, R. J. Gorte and W. E. Farneth, J. Phys. Chem., 1990, 94, 2063–2067 CrossRef CAS.
- A. Oasmaa, D. C. Elliott and J. Korhonen, Energy Fuels, 2010, 24, 6548–6554 CrossRef CAS.
- G. Fogassy, N. Thegarid, Y. Schuurman and C. Mirodatos, Energy Environ. Sci., 2011, 4, 5068 CAS.
- P. D. Chantal, S. Kaliaguine and J. L. Grandmaison, Appl. Catal., 1985, 18, 133–145 CrossRef CAS.
- T. Jin, D. H. Xia, Y. Z. Xiang and Y. L. Zhou, Pet. Sci. Technol., 2009, 27, 1821–1835 CrossRef CAS.
- S. J. Collins and P. J. Omalley, J. Catal., 1995, 153, 94–99 CrossRef CAS.
- H.-K. Min and S. B. Hong, J. Phys. Chem. C, 2011, 115, 16124–16133 CAS.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.