Synchronously synthesized core–shell LiNi1/3Co1/3Mn1/3O2/carbon nanocomposites as cathode materials for high performance lithium ion batteries†
Tao
Mei
a,
Yongchun
Zhu
*a,
Kaibin
Tang
a and
Yitai
Qian
*ab
aHefei National Laboratory for Physical Science at the Microscale and Department of Chemistry, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China
bSchool of Chemistry and Chemical Engineering, Shandong University, Jinan 250100, PR China. E-mail: ytqian@ustc.edu.cn; Fax: +86-551-360-7402; Tel: +86-551-360-1589
First published on 8th October 2012
Abstract
LiNi1/3Co1/3Mn1/3O2/carbon core–shell nanocomposites with sizes of ∼100 nm and carbon shell thicknesses of ∼6 nm are obtained by a modified Pechini process, in which LiNi1/3Co1/3Mn1/3O2 is formed synchronously with a carbon coating in the presence of polyethylene glycol-600. Electrochemical measurements show that the nanocomposites deliver a stable discharge capacity of 175 mA h g−1 at 1 C and a capacity decay rate of <3% after 100 cycles. The effects of synthesis temperature on the electrochemical performance of the nanocomposites are examined, which shows that the discharge capacities increase from 154 to 175 mA h g−1 as the temperature increases from 800 to 1000 °C. Meanwhile, the electrochemical performances of the nanocomposites with carbon content varying from 0 to 20.8% are examined. Among these composites, that with 15.5% carbon content exhibits the highest and most stable discharge behaviour at 1 C for 100 cycles.
Introduction
As one of the most promising cathode materials, LiNi1/3Co1/3Mn1/3O2 (LNCMO) has attracted considerable interest1–7 due to its high theoretical capacity, good thermal stability, high level of safety and lack of significant change during charge/discharge cycles.8 However, the delivered capacities have been shown to decrease when a high rate current density is applied.9 It is known that lithium ion battery electrodes store and release electrical energy by the insertion and extraction of Li+ ions and electrons throughout the electrode material. However, for the bulk LNCMO electrode, lithium ion diffusion is kinetically hindered. As a result of the low Li+ diffusion coefficient, the degree of polarization and aggregation in the sample increases with each charge–discharge cycle and ultimately results in poor rate capability; hence, the electrode materials used in lithium ion batteries must allow rapid ionic and electronic diffusion in order to achieve high rate performances. Usually, nanocrystallization technology 10 and carbon-coating 11 are used to improve the electrochemical performance of LNCMO cathode materials.The particle size, crystallinity, homogeneity and electrochemical performance of LNCMO is strongly dependent on the synthesis route.12,13 As solid state reaction methods usually result in large particles with irregular morphologies,14,15 different synthesis methods have been developed to prepare LNCMO particles, which include the molten salt method,16–18 the co-precipitation method,19,20 and so on. General synthesis methods of LNCMO usually involve first obtaining the mixed hydroxide precursor (Ni1/3Mn1/3Co1/3)(OH)2 by a co-precipitation method, and then calcining with a lithium compound such as LiOH. Carbonate co-precipitation was used to synthesize LNCMO at 750–900 °C, and the obtained products with sizes of around 8 μm delivered a discharge capacity of over 143 mA h g−1 at 2.5 C.19 The obtained (Ni1/3Co1/3Mn1/3)(OH)2 and LiOH·H2O powders were first heated at 480 °C for 5 h, and then calcined at 1000 °C for 10 h. The as-obtained LNCMO with sizes of about 10 μm retained a discharge capacity of 172 mA h g−1 after 50 cycles under a current density of 20 mA g−1.20
Since integration of Li into the matrix does not have to take place by a solid-state process in the calcination step, other methods of including lithium in one single precursor should be advantageous in obtaining relatively small particles such as the polymer template route,21–23 the supercritical water method,24 the electro-spinning method,25 the use of a microemulsion,26 the sol–gel method,27–29 and the hydrothermal route.30,31 LNCMO with particle sizes ranging from 0.5 to 2 μm were prepared by both the spray dry method and the metal acetate decomposition method. The reversible capacity after 35 cycles was 166 mA h g−1 at a current density of 0.2 mA cm−1.32 LNCMO (300–500 nm) was also synthesized by a hydrothermal method followed by a calcination process at 850 °C for 6 h and exhibited a discharge capacity of 187.7 mA h g−1 at 20 mA g−1 and maintained 97.9% of its capacity at the end of 40 cycles at 1.0 C.31 The Pechini method is a wet chemical technique that is easy to perform and yields oxides with good chemical homogeneity. Therefore the calcination time can be kept relatively short, which minimizes the problem of the loss of lithium through evaporation.33,34 LNCMO powders were also synthesized by the Pechini method. The material calcined at 1000 °C exhibited a capacity of around 180 mA h g−1 and maintained its capacity over 50 cycles at C/5.34 Similarly, LNCMO with a particle size of about 3.0 μm was prepared at 1000 °C, and retained a capacity of 124 mA h g−1 after 50 cycles at 1 C.33
Meanwhile, carbon-coating was also used to improve cycling performance and rate capability.11 Carbon coating could significantly improve the electrochemical performance of active materials by providing high electronic conductivity, good lithium permeability, and flexible accommodation of volume change.11 Much research has confirmed that carbon-coating can enhance the battery’s high rate performance.35 Usually, carbon-coated LNCMO is prepared through the formation of LNCMO followed by coating by carbon with a calcining carbon source. However, this method usually results in an uneven coating.36,37 Carbon-coated LNCMO obtained by calcining LNCMO with citric acid exhibited a discharge capacity of 186 mA h g−1 at 0.2 C. After the 100th cycle, the cell delivered about 92.4% of the initial discharge capacity.36 Li[Li0.2Mn0.54Ni0.13Co0.13]O2/carbon nanocomposites obtained by a slurry mixing technique exhibited a capacity of 150 mA h g−1 at 2 C with a capacity retention of 98% over 30 cycles.37
In this study, we demonstrate a synchronous carbon-coating technology to synthesize LiNi1/3Co1/3Mn1/3O2/carbon (C-LNCMO) nanocomposites. LNCMO particles were evenly coated by carbon to form a core–shell structure. The C-LNCMO nanocomposites demonstrated a stable discharge capacity of 170 mA h g−1 after 100 cycles at 1 C. It was found that the electrochemical properties of the samples were highly dependent on the synthesis temperature and the carbon content. Such an interesting core–shell nanostructure resulted in a high rate performance for both cycling stability and capability.
Experimental
Preparation of C-LNCMO nanocomposites
In a typical synthesis process, stoichiometric amounts (with 10% excess lithium) of metal nitrates, LiNO3, Ni(NO3)2·6H2O, Co(NO3)2·6H2O and Mn(NO3)2·4H2O of analysis grade, corresponding to 0.06 mol of LNCMO, were dissolved in 100 ml of distilled water (solution A). Citric acid and ethylene glycol were used as the monomers for the formation of the polymeric matrix; 0.12 mol of citric acid and 0.48 mol of ethylene glycol was dissolved in 100 ml of ethanol (solution B). Solution A was added in a drop-wise manner to solution B under magnetic stirring to obtain solution C (= A + B), then a moderate amount of polyethylene glycol-600 (PEG-600) was added to solution C. The polymeric gels were first stirred for 2 h and then calcined in air at 80 °C for 6 h, followed by heat-treatment in an electric furnace heated to 1000 °C at 20 °C min−1 for 4 h, in which nitrogen was used as a background gas at a constant feed rate of 60 ml min−1. While cooling down after calcination, the as-obtained products were collected for further characterization.Preparation of LNCMO nanocomposites
For comparison, LNCMO nanocomposites were prepared without using PEG-600 while the other conditions remained the same.Characterization of C-LNCMO nanocomposites
X-ray powder diffraction (XRD) patterns of the products were recorded on a Philips X’pert X-ray diffractometer with Cu Kα radiation (k = 1.54182 Å). The microstructure was observed on a transmission electron microscope (TEM, H7650) with an attached energy dispersive X-ray (EDX) spectroscopy system, a high-resolution transmission electron microscope (HRTEM, JEOL-2010) with an accelerating voltage of 200 kV and a field-emitting scanning electron microscope (SEM, JEOL-JSM-6700F). Raman spectroscopy was carried out on a JY LABRAM-HR confocal laser micro-Raman spectrometer using Ar+ laser excitation with a wavelength of 514.5 nm. Elemental carbon analysis of C-LNCMO was performed by a C-S 600 Determinator (Eltar, Germany).Charge/discharge tests were carried out using coin-type cells (size: 2016), which consisted of an active material working electrode and a Li foil counter electrode separated by a Celgard 2300 microporous membrane. To prepare the working electrode, the active material, super P carbon black, and polyvinylidene fluoride (PVDF) were mixed in the weight ratio 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:10 with N-methyl pyrrolidone (NMP) serving as the solvent to obtain a slurry. After coating the slurry onto an aluminum foil, the electrode was dried at 120 °C in a vacuum for 12 h. Typical loadings of the electrodes were between 3 and 4 mg cm−2 of the active material. A 1 mol L−1 solution of LiPF6 dissolved in ethylene carbonate/dimethyl carbonate (EC/DMC) (1:1 volume ratio) was used as the electrolyte. The cells were assembled in an argon-filled glove box (Mikrouna, Super 1220/750/900, China). The cut-off voltages were 2.8–4.6 V. The final capacities were calculated based on weight of the LNCMO.
:10:10 with N-methyl pyrrolidone (NMP) serving as the solvent to obtain a slurry. After coating the slurry onto an aluminum foil, the electrode was dried at 120 °C in a vacuum for 12 h. Typical loadings of the electrodes were between 3 and 4 mg cm−2 of the active material. A 1 mol L−1 solution of LiPF6 dissolved in ethylene carbonate/dimethyl carbonate (EC/DMC) (1:1 volume ratio) was used as the electrolyte. The cells were assembled in an argon-filled glove box (Mikrouna, Super 1220/750/900, China). The cut-off voltages were 2.8–4.6 V. The final capacities were calculated based on weight of the LNCMO.
Results and discussion
Fig. 1a and b show typical XRD patterns of the products without and with the carbon coating prepared at 1000 °C, respectively. The diffraction peaks in Fig. 1a and b could be readily indexed as the hexagonal α-NaFeO2 structure with a space group of R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m.4,6 The well defined XRD patterns display the hexagonal doubles (006)/(012) and (108)/(110) with a clear split, which indicates that the samples have good hexagonal ordering and good layered characteristics.38 The chemical composition of the as-obtained sample prepared at 1000 °C was determined by ICP-OES (Table 1). It is seen that the final ratio of Li:(Ni + Co + Mn) is larger than 1.0, which could be considered an achievement of the Pechini method for the relatively short calcining time.34 In fact, studies have shown that a lithium excess promotes the electrochemical performance of the LNCMO material.39,40 The c/a ratio and the I(003)/I(104) intensity ratio are also shown in Table 1. The c/a ratio is an indicator of the cation ordering in the layered structure.41 The larger the ratio, the lower the cation disorder. The sample synthesized at 1000 °C exhibits a ratio of 4.974. The intensity ratio of the (003) peak to the (104) peak is also used to examine the crystallinity of the LNCMO powders.5 The higher the I(003)/I(104) value, the better the layered structure of the LNCMO powder. It should be noted that the intensity ratio I(003)/I(104) is 1.2, indicating low cation mixing.42,43
m.4,6 The well defined XRD patterns display the hexagonal doubles (006)/(012) and (108)/(110) with a clear split, which indicates that the samples have good hexagonal ordering and good layered characteristics.38 The chemical composition of the as-obtained sample prepared at 1000 °C was determined by ICP-OES (Table 1). It is seen that the final ratio of Li:(Ni + Co + Mn) is larger than 1.0, which could be considered an achievement of the Pechini method for the relatively short calcining time.34 In fact, studies have shown that a lithium excess promotes the electrochemical performance of the LNCMO material.39,40 The c/a ratio and the I(003)/I(104) intensity ratio are also shown in Table 1. The c/a ratio is an indicator of the cation ordering in the layered structure.41 The larger the ratio, the lower the cation disorder. The sample synthesized at 1000 °C exhibits a ratio of 4.974. The intensity ratio of the (003) peak to the (104) peak is also used to examine the crystallinity of the LNCMO powders.5 The higher the I(003)/I(104) value, the better the layered structure of the LNCMO powder. It should be noted that the intensity ratio I(003)/I(104) is 1.2, indicating low cation mixing.42,43
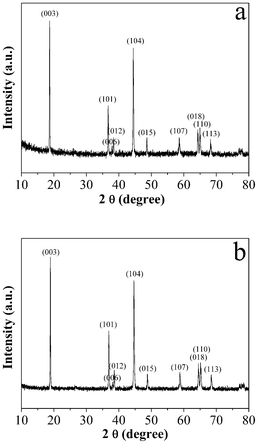 | ||
| Fig. 1 Typical XRD patterns of the products prepared at 1000 °C: (a) LNCMO and (b) C-LNCMO nanocomposites. | ||
| Temperature/°C | Composition | Li:(Ni + Co + Mn) |
c/a | I(003)/I(104) |
|---|---|---|---|---|
| 800 | Li1.06Mn0.34Ni0.32Co0.34O2 | 1.06 | 4.971 | 1.21 |
| 900 | Li1.04Mn0.34Ni0.33Co0.33O2 | 1.04 | 4.973 | 1.20 |
| 1000 | Li1.03Mn0.34Ni0.33Co0.33O2 | 1.03 | 4.974 | 1.20 |
To investigate the nature of the carbon, Raman microprobe spectroscopy was employed. The Raman spectra of LNCMO and C-LNCMO are shown in Fig. 2. Both spectra have a broad band between 400 and 650 cm−1 comprising bands at 474, 554 (Ni–O), 486, 595 (Co–O), and 592 cm−1 (Mn–O), which represent the vibrations within a hexagonal lattice belonging to the same space symmetry group (Rm).44 In the case of C-LNCMO, there are two bands at 1356 and 1587 cm−1 which are typical bands of carbon nominated as the D band and G band, corresponding to the vibration of carbon atoms with dangling bonds at the in-plane terminations of disordered graphite and the vibration of sp2-bonded carbon atoms in a 2D hexagonal lattice, respectively.45 The ID/IG value of the sample was 0.92, which indicates that the as-obtained sample possessed a large amount of disorder and a large number of defects.46
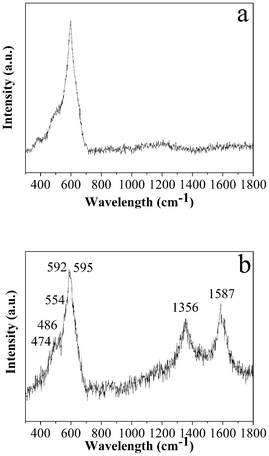 | ||
| Fig. 2 Typical Raman spectra of the products prepared at 1000 °C: (a) LNCMO and (b) C-LNCMO nanocomposites. | ||
The morphologies and structures of the as-prepared samples were further analyzed by SEM and TEM. Fig. 3a and b show typical SEM images of the as-obtained C-LNCMO prepared at 1000 °C. We could clearly see that the particle size of the C-LNCMO was around 100 nm. From the TEM images (Fig. 3c and d), the core–shell structure could be observed. The LNCMO nanoparticles were uniformly coated by carbon layers. The results from the observation of the SEM and TEM images indicated that these C-LNCMO nanoparticles had smooth surfaces, uniform shapes and a tendency to interconnect with each other. The HRTEM image in Fig. 3e further confirmed that the structure was constructed from an LNCMO core and a carbon shell. The strong contrast between the light edge and dark center of the nanocomposite provided evidence of its core–shell structure. The lattice spacing was about 4.63 Å, corresponding to the lattice spacing of the (003) plane for LNCMO. It could be seen that the thickness of the carbon shell was about 6 nm. The results from the EDS analysis for the C-LNCMO core–shell nanocomposites are shown in Fig. 3f. The molar ratio of the elements Ni, Co and Mn was approximately the same as the intended ratio of 1:1:1.
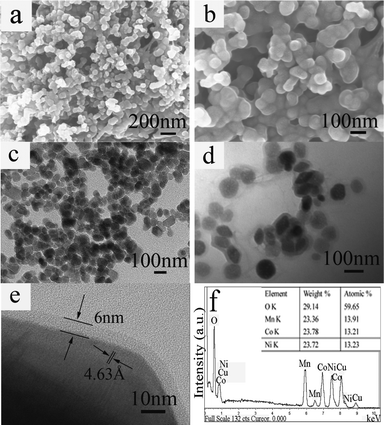 | ||
| Fig. 3 (a, b) Typical SEM images, (c, d) TEM images, (e) HRTEM image, (f) EDS spectrum of C-LNCMO prepared at 1000 °C. | ||
The formation process of the core–shell structure could be described as follows. Initially, PEG could serve as a complexing agent and react with the metal ions to form “metal ion–PEG” complexes.47,48 Then, as the temperature gradually rises, the glycol-type molecules might dehydrate to form polymer-like organic components, which is similar to a previous study.49 At the same time, the recrystallization of the LNCMO nanocrystals could densify the core and produce a vacant region between the outer shells. Meanwhile, the polymer-like organic components (or slightly carbonized polymers) are further converted into the carbon shell. As a result, the C-LNCMO nanocrystals are formed from the “metal ion-PEG” complexes gradually. It should be noted that unlike the previous literature on preparation at 1000 °C,33,34 the average sizes of the samples prepared by this modified Pechini process were significantly decreased. On one hand, when the PEG-600 served as a dispersant, the surface tension was expected to reduce significantly, and the sol particle sizes were expected to decrease correspondingly.50 On the other hand, the calcination time could be kept relatively short due to the fact that the Pechini method was a wet chemical technique which was easy to perform and yielded oxides with good chemical homogeneity.
In order to analyze the electrochemical properties of the C-LNCMO in detail, comparative experiments were carried out. The chemical composition and c/a and I(003)/I(104) (intensity) ratios for samples obtained at different temperatures are shown in Table 1. Fig. 4a shows the life cycle performance of the as-obtained C-LNCMO at different temperatures. It can be seen that all the samples exhibited good cycle stability, however, the discharge capacities increased from 154 to 175 mA h g−1 as the synthesis temperature increased from 800 to 1000 °C, which is in agreement with the literature.33,34 Since the material calcined at 1000 °C showed the highest capacity, it was selected for further investigation. Fig. 4b shows the first charge–discharge cycles at 1 and 5 C of C-LNCMO prepared at 1000 °C. It can be seen that the charge/discharge behavior of the C-LNCMO was different to that of the layered LiCoO2, which had a well-defined plateau at about 3.9 V.51 On applying the current, the cell voltage rapidly increased to about 3.7 V and then gradually increased until it reached the upper voltage limit of 4.6 V for both rates. This charge/discharge behavior of C-LNCMO agrees well with the reports by other groups.52,53Fig. 4c shows the results from a cycling experiment in which the discharge rate was increased in steps from 1 C to 5 C, with five cycles at each current. The capacity decreases somewhat with increasing current, but the cell managed to deliver around 130 mA h g−1 even for 5 C. When the rate was returned to 1 C, the capacity still remained at 170 mA h g−1. Fig. 4d shows the cycle performances of the as-obtained C-LNCMO prepared at 1000 °C for different charge/discharge rates, at 2, 3 and 5 C, respectively (the first five cycles were at 0.1 C). After the 50th cycle, it could be seen that the discharge capacities were 97.4, 95.6 and 88.1% of those in the 6th cycle for 2, 3 and 5 C, respectively. Compared with nonsynchronous synthesized C-LNCMO,36 this result shows that the synchronous synthesized C-LNCMO exhibited a superior performance.
 | ||
| Fig. 4 (a) The life cycle performance of the as-obtained C-LNCMO at different temperatures, (b) typical first charge/discharge behavior of the C-LNCMO at 1 and 5 C, (c) cycling of the C-LNCMO calcined at 1000 °C. The discharge rate was varied from 1 C to 5 C. The charge rate was kept constant at C/5 throughout, (d) cycle performance of the C-LNCMO composite at 2, 3 and 5 C (the first five cycles were at 0.1 C). | ||
The cycle performances of C-LNCMO prepared at 1000 °C with various carbon contents are shown in Fig. 5. The initial discharge capacity of the LNCMO at 1 C was about 162 mA h g−1. Compared with the pristine sample, the C-LNCMO exhibited a comparable discharge capacity of 175 mA h g−1. After the 100th cycle at 1 C, the cell delivered about 85.3% and 97.8% of the initial discharge capacity for the pristine LNCMO and the C-LNCMO, respectively. It is proposed that the enhanced electrochemical properties due to the carbon-coating are attributed to increased electronic conductivity because the carbon distributed among the surfaces of the LNCMO powder favored the transfer of electrons and reduced cell polarization.36 After carbon-coating, the electronic conductivity was increased, which reduced the cell polarization and prevented the evolution of oxygen from the cathodes at the end of charge, thus suppressing the reduction in the capacity, especially at higher rates. The discharge capacity increased from 159 to 171 mA h g−1 when the carbon content increased from 10.4 to 15.5%. However, the C-LNCMO composite with a carbon content of 20.8% delivered a discharge capacity of 163 mA h g−1, which was less than that of the C-LNCMO composite with a carbon content of 15.5%. This could be attributed to the coating layer being too dense to facilitate easy lithium-ion diffusion.37 This result, indicating the optimal carbon content required to improve the electrochemical properties, is consistent with that for Li2FeSiO4.54
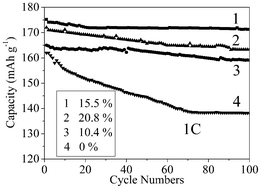 | ||
| Fig. 5 The cycle performances of C-LNCMO prepared at 1000 °C with various carbon contents. | ||
The special core–shell nanostructure results in an improvement in both the cycling performance and the capability at higher rates, which can be mainly attributed to the following: first, the particle size of the C-LNCMO was around only 100 nm. From the literature, it can be seen that nanocrystallization technology is valid for improving the electrochemical performance of LNCMO cathode materials.10 Secondly, for our C-LNCMO materials, each LNCMO nanoparticle was uniformly coated by carbon layers. It has been reported that in order to minimize the polarisation due to the insertion process, the carbon must be uniformly distributed around each active particle.55,56 The uniformity of the carbon distribution on the framework could relax the strain stress, buffer the volume expansion and hence improve the cycle stability of the materials. Finally, not only nanocrystallization and carbon coating, but also the construction of nanocomposites with high conductivities and activities have been used to improve the electrochemical performance of lithium ion batteries.57,58 PEG-600 played a critical role in the nano-core–shell structure formation, which not only improved the dispersion of the LNCMO without aggregation, but also directed the growth of small LNCMO nanoparticles and the development of the network nanostructure. Such a self-assembled three-dimensional network nanostructure exhibits a high conductivity, a large specific surface area and a unique porosity and could be used as the conductive additive and the supporting matrix. The graphitic shells and the interconnected carbon-framework structure ensured the fast and continuous transportation of electrons in the electrode, and provided continuous paths between the LNCMO and the carbon shell, which was favorable for electrons moving unimpeded over the LNCMO particles to attain a high rate capability.59
Conclusions
LiNi1/3Co1/3Mn1/3O2/carbon core–shell nanocomposites with sizes of around 100 nm were prepared by a synchronous carbon-coating technology. PEG-600 was used as the carbon source and the dispersing agent. The C-LNCMO nanocomposites were prepared at 1000 °C and the carbon content was 15.5%. They demonstrated a stable discharge capacity of 175 mA h g−1 at 1 C and exhibited a capacity decay rate of <3% after 100 cycles. Since the Pechini method could be well suited for the preparation of materials such as spinel LiMn2O4,60,61 Li2MnSiO4,62 LiNi0.5Co0.5VO463 and so on for lithium-ion batteries, further studies to use this modified synchronous carbon-coating technology to obtain evenly carbon coated core–shell structures and improve both the cycling performance and the rate capability are in progress.Acknowledgements
This work was financially supported by the National Nature Science Fund of China (No. 91022033) and the 973 Project of China (No. 2011CB935901), Anhui Provincial Natural Science Foundation (1208085QE101) and the Fundamental Research Funds for the Central Universities (No. WK 2340000027).References
- Y. Huang, J. Chen, J. Ni, H. Zhou and X. Zhang, J. Power Sources, 2009, 188, 538 CrossRef CAS.
- X. Luo, X. Wang, L. Liao, S. Gamboa and P. Sebastian, J. Power Sources, 2006, 158, 654 Search PubMed.
- N. Yabuuchi and T. Ohzuku, J. Power Sources, 2003, 119, 171 CrossRef.
- I. Belharouak, Y. K. Sun, J. Liu and K. Amine, J. Power Sources, 2003, 123, 247 CrossRef CAS.
- K. M. Shaju, G. V. S. Rao and B. V. R. Chowdari, Electrochim. Acta, 2002, 48, 145 CrossRef CAS.
- T. Ohzuku and Y. Makimura, Chem. Lett., 2001, 30, 642 CrossRef.
- Z. H. Lu, D. D. MacNeil and J. R. Dahn, Electrochem. Solid-State Lett., 2001, 4, A200 CrossRef CAS.
- Z. D. Huang, X. M. Liu, S. W. Oh, B. Zhang, P. C. Ma and J. K. Kim, J. Mater. Chem., 2011, 21, 10777 RSC.
- G. Kim, J. Kim, S. Myung, C. Yoon and Y. Sun, J. Electrochem. Soc., 2005, 152, A1707 CrossRef CAS.
- M. Kunduraci, J. F. Al-Sharab and G. G. Amatucci, Chem. Mater., 2006, 18, 3585 CrossRef CAS.
- Z. Chen, Y. Qin, K. Amine and Y. K. Sun, J. Mater. Chem., 2010, 20, 7606 RSC.
- P. He, H. R. Wang, L. Qi and T. Osaka, J. Power Sources, 2006, 160, 627 CrossRef CAS.
- L. Q. Zhang, X. Q. Wang, T. Muta, D. C. Li, H. Noguchi, M. Yoshio, R. Z. Ma, K. Takada and T. Sasaki, J. Power Sources, 2006, 162, 629 Search PubMed.
- M. Kageyama, D. Li, K. Kobayakawa, Y. Sato and Y. S. Lee, J. Power Sources, 2006, 157, 494 CrossRef CAS.
- L. Tan and H. Liu, Solid State Ionics, 2010, 181, 1530 Search PubMed.
- M. V. Reddy, G. V. S. Rao and B. V. R. Chowdari, J. Power Sources, 2006, 159, 263 CrossRef CAS.
- K. Du, Z. Peng, G. Hu, Y. Yang and L. Qi, J. Alloys Compd., 2009, 476, 329 CrossRef CAS.
- Z. Chang, Z. Chen, F. Wu, X. Z. Yuan and H. Wang, Electrochim. Acta, 2009, 54, 6529 CrossRef CAS.
- T. H. Cho, S. M. Park, M. Yoshio, T. Hirai and Y. Hideshima, J. Power Sources, 2005, 142, 306 CrossRef CAS.
- Y. K. Sun, M. H. Lee, Y. Kang and S. T. Myung, Electrochim. Acta, 2004, 50, 939 CrossRef CAS.
- C. Ding, Q. Meng, L. Wang and C. Chen, Mater. Res. Bull., 2009, 44, 492 CrossRef CAS.
- K. M. Shaju and P. G. Bruce, Adv. Mater., 2006, 18, 2330- Search PubMed.
- N. N. Sinha and N. Munichandraiah, J. Electrochem. Soc., 2010, 157, A647 CrossRef CAS.
- J. W. Lee, J. H. Lee, T. T. Viet, J. Y. Lee, J. S. Kim and C. H. Lee, Electrochim. Acta, 2010, 55, 3015 CrossRef CAS.
- Y. Ding, P. Zhang, Z. Long, Y. Jiang and D. Gao, J. Alloys Compd., 2008, 462, 340 CrossRef CAS.
- C. H. Lu and Y. K. Lin, J. Power Sources, 2009, 189, 40 CrossRef CAS.
- M. Sathiya, A. Prakash, K. Ramesha and A. Shukla, Mater. Res. Bull., 2009, 44, 1990 Search PubMed.
- C. H. Chen, C. J. Wang and B. J. Hwang, J. Power Sources, 2005, 146, 626 CrossRef CAS.
- R. Santhanam and B. Rambabu, J. Power Sources, 2010, 195, 4313 CrossRef CAS.
- S. T. Myung, M. H. Lee, S. Komaba, N. Kumagai and Y. K. Sun, Electrochim. Acta, 2005, 50, 4800 CrossRef CAS.
- F. Wu, M. Wang, Y. Su, L. Bao and S. Chen, J. Power Sources, 2010, 195, 2362 CrossRef CAS.
- H. Noguchi, D. C. Li, T. Muta, L. Q. Zhang and M. Yoshio, J. Power Sources, 2004, 132, 150 CrossRef CAS.
- H. Xia, H. L. Wang, W. Xiao, L. Lu and M. O. Lai, J. Alloys Compd., 2009, 480, 696 CrossRef CAS.
- P. Samarasingha, D. H. Tran-Nguyen, M. Behm and A. Wijayasinghe, Electrochim. Acta, 2008, 53, 7995 CrossRef CAS.
- H. Huang, S. C. Yin and L. Nazar, Electrochem. Solid-State Lett., 2001, 4, A170 CrossRef CAS.
- Z. Y. Wen, B. Lin, X. Y. Wang and Y. Liu, J. Solid State Electrochem., 2010, 14, 1807 CrossRef CAS.
- A. Manthiram, J. Liu, Q. Y. Wang and B. Reeja-Jayan, Electrochem. Commun., 2010, 12, 750 CrossRef CAS.
- P. Periasamy, H. S. Kim, S. H. Na, S. I. Moon and J. C. Lee, J. Power Sources, 2004, 132, 213 CrossRef CAS.
- J. Choi and A. Manthiram, Electrochem. Solid-State Lett., 2005, 8, C102 CrossRef CAS.
- Y. M. Todorov and K. Numata, Electrochim. Acta, 2004, 50, 495 Search PubMed.
- T. Ohzuku, K. Nakura and T. Aoki, Electrochim. Acta, 1999, 45, 151 CrossRef CAS.
- D. C. Li, T. Muta, L. Q. Zhang, M. Yoshio and H. Noguchi, J. Power Sources, 2004, 132, 150 CrossRef CAS.
- T. Ohzuku, A. Ueda, M. Nagayama, Y. Iwakoshi and H. Komori, Electrochim. Acta, 1993, 38, 1159 CrossRef CAS.
- M. Kerlau, M. Marcinek, V. Srinivasan and R. M. Kostecki, Electrochim. Acta, 2007, 53, 1385 Search PubMed.
- D. G. Mcculloch, S. Prawer and A. Hoffman, Phys. Rev. B: Condens. Matter, 1994, 50, 5905 CrossRef CAS.
- T. Mei, T. Li, H. Y. Bi, L. B. Wang, Y. C. Zhu and Y. T. Qian, Eur. J. Inorg. Chem., 2010, 4314 Search PubMed.
- W. Y. Yin, J. Su, M. H. Cao, C. Y. Ni, C. W. Hu and B. Q. Wei, J. Phys. Chem. C, 2010, 114, 65 CrossRef CAS.
- J. Yang, C. X. Li, Z. W. Quan, D. Y. Kong, X. M. Zhang, P. P. Yang and J. Lin, Cryst. Growth Des., 2008, 8, 695 CrossRef CAS.
- X. Liang, B. A. Xu, S. M. Kuang and X. Wang, Adv. Mater., 2008, 20, 3739 CrossRef CAS.
- Y. F. Zhang, J. X. Zhang, Q. M. Lu and Q. Y. Zhang, Mater. Lett., 2006, 60, 2443 Search PubMed.
- H. Xia, L. Lu, Y. S. Meng and G. Ceder, J. Electrochem. Soc., 2007, 154, A337 Search PubMed.
- J. Liu, W. Qiu, L. Yu, G. Zhang, H. Zhao and T. Li, J. Power Sources, 2007, 174, 701 Search PubMed.
- Y. J. Shin, W. J. Choi, Y. S. Hong, S. Yoon, K. S. Ryu and S. H. Chang, Solid State Ionics, 2006, 177, 515 Search PubMed.
- H. J. Guo, K. X. Xiang, X. Cao, X. H. Li, Z. X. Wang and L. M. Li, Trans. Nonferrous Met. Soc. China, 2009, 19, 166 Search PubMed.
- R. Dominko, M. Gaberscek, J. Drofenik, M. Bele, S. Pejovnik and J. Jamnik, J. Power Sources, 2003, 119, 770 CrossRef.
- R. Dominko, M. Gaberscek, J. Drofenik, M. Bele and J. Jamnik, Electrochim. Acta, 2003, 48, 3709 CrossRef CAS.
- C. X. Guo, M. Wang, T. Chen, X. W. Lou and C. M. Li, Adv. Energy Mater., 2011, 1, 736 Search PubMed.
- C. X. Guo, Y. Q. Shen, Z. L. Dong, X. D. Chen, X. W. Lou and C. M. Li, Energy Environ. Sci., 2012, 5, 6919 RSC.
- F. D. Han, Y. J. Bai, R. Liu, B. Yao, Y. X. Qi, N. Lun and J. X. Zhang, Adv. Energy Mater., 2011, 1, 798 Search PubMed.
- W. Liu, K. Kowal and G. C. Farrington, J. Electrochem. Soc., 1996, 143, 3590 CAS.
- W. Liu, G. C. Farrington, F. Chaput and B. Dunn, J. Electrochem. Soc., 1996, 143, 879 CAS.
- R. Dominko, M. Bele, M. Gaberscek, A. Meden, M. Remskar and J. Jamnik, Electrochem. Commun., 2006, 8, 217 CrossRef CAS.
- S. Vivekanandhan, M. Venkateswarlu and N. Satyanarayana, Mater. Chem. Phys., 2005, 91, 54 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ra21392c |
| This journal is © The Royal Society of Chemistry 2012 |