High-performance CO2 sorbents from algae†
Marta
Sevilla
*a,
Camillo
Falco
bc,
Maria-Magdalena
Titirici
c and
Antonio B.
Fuertes
a
aInstituto Nacional del Carbón (CSIC), P.O. Box 73, 33080, Oviedo, Spain. E-mail: martasev@incar.csic.es; Fax: +34 985 29 76 62; Tel: +34 985 11 90 90
bIASS – Institute for Advanced Sustainability Studies, Berliner Strasse 130, 14467, Potsdam Germany
cColloid Chemistry, Max-Planck Institute for Colloids and Interfaces, Am Muhlenberg 1, 14476, Potsdam Germany
First published on 25th October 2012
Abstract
Highly porous N-doped carbon materials with apparent surface areas in the 1300–2400 m2 g−1 range and pore volumes up to 1.2 cm3 g−1 have been synthesized from hydrothermal carbons obtained from mixtures of algae and glucose. The porosity of these materials is made up of uniform micropores, most of them having sizes <1 nm. Moreover, they have N contents in the 1.1–4.7 wt% range, and the heteroatom is mainly a pyridone-type structure. These microporous carbons present unprecedented large CO2 capture capacities, up to 7.4 mmol g−1 (1 bar, 0 °C). The importance of the pore size on the CO2 capture capacity of microporous carbon materials is clearly demonstrated. Indeed, a good correlation between the CO2 capture capacity at sub-atmospheric pressure and the volume of narrow micropores is observed. The results suggest that pyridinic-N, pyridonic/pyrrolic-N and quaternary-N do not contribute significantly to the CO2 adsorption capacity, owing probably to their low basicity in comparison with amines. These findings will help the design of high-performance CO2 capture sorbents.
1. Introduction
The growing environmental concerns for global warming and climate change are reflected in the increasing number of works related to the development of more-efficient and improved processes for CO2 capture and storage. Capturing CO2 is by far the most energy-expensive part of carbon capture and storage technologies. In this regard, physisorption of CO2 in porous solids using pressure, temperature or vacuum swing adsorption processes (PSA, TSA or VSA respectively) can significantly decrease the cost, as well as the environmental impact associated with the currently used technologies involving amines. A CO2 sorbent should satisfy the following requirements: (i) large CO2 uptake, (ii) high sorption rate, (iii) good selectivity between CO2 and other competing gases in the stream (i.e. N2), (iv) easy regeneration and (v) low-cost and high availability. So far, numerous porous solids have been investigated, such as zeolites, metal–organic frameworks (MOFs), polymers, porous carbons including templated carbons, activated carbons and carbide-derived carbons (CDCs), or organic-inorganic hybrid materials.1–6 Among them, porous carbons stand out in terms of cost, stability, availability, large surface area, easy-to-design pore structure and low energy requirements for regeneration. More specifically, activated carbons continue to be the primary choice for many adsorption-based processes due to their cost-availability and microstructure. In this regard, research on novel activation procedures which allow better control over the microstructure and PSD is crucial for further development of many adsorption-based systems, such as hydrogen storage, supercapacitors or CO2 capture. However, not only the activation conditions influence the microstructure of the final material, but also the precursor used. For a large-scale sustainable production, naturally grown materials should be used as precursors. In this respect, biomass is highly attractive. More specifically, microalgae stand out for very high photosynthesis efficiency and are considered to be a fast growing biomass, commonly doubling their mass within a 24 h growth window,7–10 which would ensure the quick availability of this precursor. An additional advantage of microalgae is that some species are rich in proteins and therefore have relatively high nitrogen contents, which may allow the synthesis of N-doped materials. The synthesis of this kind of material is receiving increasing attention as the presence of nitrogen gives the carbon material improved thermal/oxidation stability and higher electrical conductivity.11,12 Furthermore, nitrogen-containing functionalities are responsible for an increase in the surface polarity of carbon and basicity,13 which could enhance the interaction between carbon and acid molecules, such as H2S, SO2 or CO2. In the present work, we will prove the efficient transformation of a microalgae-derived N-doped hydrothermal carbon into highly microporous N-doped carbon materials. In addition, we analyzed their CO2 capture capacity, and found that those microporous carbons presented unprecedented large CO2 uptakes for carbon materials, up to 7.4 mmol g−1 at 0 °C and 1 bar. Their capture capacity has been correlated with their chemical and textural characteristics, showing that the volume of narrow micropores is the parameter governing the adsorption of CO2 at sub-atmospheric conditions.2. Experimental
2.1. Preparation of activated carbons
N-doped hydrothermal carbon materials (HTC carbon) were obtained by hydrothermal co-carbonization of microalgae (Spirulina platensis) and glucose, with a mass ratio microalgae/glucose = 1.5, at a temperature of 200 °C for 24 h. Afterwards, the hydrothermal carbons were chemically activated in a vertical furnace using potassium hydroxide (Sigma-Aldrich) at temperatures in the range 650–750 °C and KOH/HTC carbon mass ratio of 1, 2 or 4. The samples were denoted as AG-x-T, where x is the mass ratios KOH/HTC carbon and T the activation temperature.2.2 Sample characterization
Elemental analysis (C, H, N, S and O) was carried out on a LECO CHN-932 microanalyzer. X-Ray photoelectron spectroscopy (XPS) was carried out by means of a Thermo Scientific K-Alpha ESCA instrument using monochromatic Al-Kα radiation (hν = 1486.6 eV). Binding energies for the high-resolution spectra were calibrated by setting C 1s at 285.0 eV. SEM images were acquired on a LEO 1550/LEO GmbH Oberkochen equipped with an Everhard Thornley secondary electron and in-lens detectors. Nitrogen sorption isotherms of the carbon samples were measured at −196 °C using a Micromeritics ASAP 2020 sorptometer. The apparent surface area was calculated using the BET method; for the selection of the appropriate relative pressure range, the ISO 9277![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2010 was followed.14 The total pore volume was determined from the amount of nitrogen adsorbed at a relative pressure (P/P0) of 0.99, whereas the micropore volume, V0, and narrow micropore volume, Vn, were determined by applying the Dubinin–Radushkevich (DR) equation to the nitrogen and carbon dioxide isotherms respectively. The average pore width, L0, was calculated by applying the equation proposed by Stoeckli and Ballerini.15 The pore size distribution (PSD) was determined via a Quench Solid State Functional Theory (QSDFT) method (implemented into Quantachrome's data software), using nitrogen adsorption data, and assuming a slit-shaped pore model. This method supposes an improvement over the non-local density functional method (NLDFT), allowing one to account explicitly for the effects of surface heterogeneity, typical in activated carbons.16,17 The false gaps at around 1 and 2 nm, which are typical artifacts of the standard NLDFT model that treats the pore walls as homogeneous graphite-like plane surfaces, disappear in the QSDFT PSD.
:2010 was followed.14 The total pore volume was determined from the amount of nitrogen adsorbed at a relative pressure (P/P0) of 0.99, whereas the micropore volume, V0, and narrow micropore volume, Vn, were determined by applying the Dubinin–Radushkevich (DR) equation to the nitrogen and carbon dioxide isotherms respectively. The average pore width, L0, was calculated by applying the equation proposed by Stoeckli and Ballerini.15 The pore size distribution (PSD) was determined via a Quench Solid State Functional Theory (QSDFT) method (implemented into Quantachrome's data software), using nitrogen adsorption data, and assuming a slit-shaped pore model. This method supposes an improvement over the non-local density functional method (NLDFT), allowing one to account explicitly for the effects of surface heterogeneity, typical in activated carbons.16,17 The false gaps at around 1 and 2 nm, which are typical artifacts of the standard NLDFT model that treats the pore walls as homogeneous graphite-like plane surfaces, disappear in the QSDFT PSD.
3. Results and discussion
In a previous work, we showed that hydrothermal co-carbonization of a protein rich microalgae, i.e. Spirulina platensis, with glucose at T = 200 °C for 24 h yields a N-doped hydrothermal carbon with a N content of 7.5 wt% and lacking framework confined porosity (SBET ∼ 6 m2 g−1).18 As shown in Fig. S1a in the ESI†, this material is composed mostly of spheroidal particles with rough surfaces. After the chemical activation step, a complete change in the morphology of the particles takes place, as can be seen in Fig. S1b†. Now the material is composed of particles of irregular morphology and sharp edges with a surface apparently smooth. However, higher magnification SEM micrographs reveal a high degree of surface roughness present as well inside the particles (see Fig. S1c†), which highlights the presence of porosity. This complete rearrangement of the particles morphology suggests melting down of the precursor during the activation process. This behavior has been previously observed for hydrothermal carbons obtained from a variety of saccharides or biomass, like glucose, starch or sawdust.19 However, when more carbonized materials, i.e. materials with a more rigid structure, such as templated carbons20 or carbide-derived carbons21 are used as precursors, the structure of the initial material is partially preserved. This allows the creation of a large amount of porosity. Thus, highly porous materials with apparent surface areas in the ∼1300–2400 m2 g−1 range and pore volumes up to ∼1.2 cm3 g−1 are obtained (see Table 1). They all exhibit type I isotherms, with a more or less pronounced knee depending on the activation conditions (Fig. S2 in ESI†). As shown by their QSDFT pore size distributions depicted in Fig. 1, they are exclusively microporous (pores < 2 nm), except for the highly activated samples (650–700 °C and KOH/HTC carbon = 4), which also exhibit a small amount of mesopores up to 3 nm. Furthermore, the main pore size system is below 1 nm in all the samples, which is a very desirable feature for CO2 capture, as CO2 adsorption is strongly favored in such micropores.1,22 Comparison of the PSD of samples activated at increasing temperature for the same KOH/HTC carbon ratio reveals an increase of the average pore size, as well as a broadening of the PSD. The same effect on the PSD is caused by the increase of the KOH/HTC carbon ratio at a fixed activation temperature. This enlargement of the porosity is accompanied by an increase of both the apparent surface area and total pore volume (see Fig. S3 in ESI†). It must be mentioned that direct activation with KOH of the algae leads to its complete burn-off. Therefore, hydrothermal carbonization is a key step to the successful conversion of algae into a product of high-added value, i.e. an activated carbon with excellent CO2 capture capacity, as will be proved here.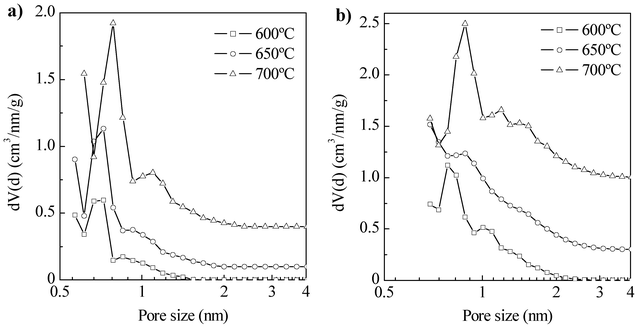 | ||
| Fig. 1 QSDFT pore size distributions of the porous carbons prepared under different activating conditions: a) KOH/HTC carbon = 2 and b) KOH/HTC carbon = 4. | ||
| Sample | BET surface area [m2 g−1] | Pore volume [cm3 g−1] | V 0 (N2) [cm3 g−1] | L 0 (N2) [nm] | V n (CO2) [cm3 g−1] | O (%) | N (%) |
|---|---|---|---|---|---|---|---|
| AG-1-700 | 1480 | 0.60 | 0.60 | 0.92 | 0.47 | 11.20 | 1.76 |
| AG-2-600 | 1260 | 0.53 | 0.51 | 0.80 | 0.47 | 18.22 | 4.65 |
| AG-2-650 | 1620 | 0.68 | 0.64 | 0.80 | 0.50 | 12.30 | 2.68 |
| AG-2-700 | 1940 | 0.82 | 0.76 | 0.92 | 0.47 | 11.00 | 1.47 |
| AG-4-600 | 1990 | 0.89 | 0.76 | 1.2 | 0.41 | 15.26 | 1.36 |
| AG-4-650 | 2180 | 1.01 | 0.87 | 1.5 | 0.36 | 16.40 | 1.12 |
| AG-4-700 | 2390 | 1.15 | 0.96 | 1.8 | 0.34 | 9.97 | 1.09 |
The results of the elemental analysis gathered in Table 1 show that the KOH activation leads to the partial loss of nitrogen contained in the precursor, from 7.5 wt% in the hydrothermal carbon to 1.1–4.7 wt% in the activated carbons. The preferential removal of N during the activation process has been already reported by other authors for other nitrogen-containing precursors.23–25 The nature of the nitrogen moieties on the carbon surface was analyzed by XPS, results shown in Fig. 2a for a representative sample, i.e. AG-2-650. Three main contributions are visible in the high-resolution N 1s: i) pyridinic-N at 398.8 eV, ii) pyrrolic-/pyridonic-N at 400.1 eV and iii) quaternary-N at 401.5 eV. Although pyrrolic-N and pyridonic-N are indistinguishable by XPS, taking into account the conditions of the activation process, e.g. oxidative environment and high temperatures (>600 °C), the peak at 400.1 eV is most likely attributed to pyridone-type structures.26 We would like to stress the fact that in all the prepared porous carbons, the pyridone-type structures are the main type of N-functionalities. These N-moieties give Lewis basicity to the materials, which might be relevant for the capture of acidic gases such as CO2.
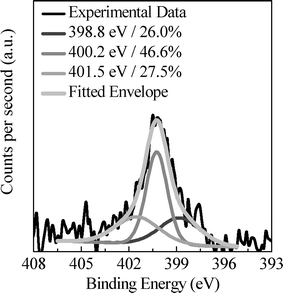 | ||
| Fig. 2 N 1s XPS spectra of AG-2-650. | ||
These microporous carbons were investigated as CO2 sorbents at sub-atmospheric conditions (up to 1 bar), typical for post-combustion capture. The results are summarized in Table 2. All the materials exhibit excellent CO2 capture capacities, higher than 5.9 mmol CO2 g−1 (260 mg CO2 g−1 or 26%) at 0 °C and 1 bar. Especially remarkable are the values obtained for AG-2-650 and AG-2-700, i.e. 7.0 and 7.4 mmol CO2 g−1 respectively, which are among the highest ever reported for porous carbon materials (see Table 3). Only Presser et al. reported a value of 7.1 mmol CO2 g−1 at 0 °C and 1 bar for a carbide-derived carbon (CDC),1 and Wahby et al. a value of 8.6 mmol CO2 g−1 (0 °C) for a petroleum pitch-derived activated carbon.27 Even at 50 °C, the CO2 capture capacity of such sustainable carbon materials is significant, in the 2.2 to 2.8 mmol CO2 g−1 range. As expected for an exothermic process, the amount of CO2 adsorbed decreases with the increase in adsorption temperature. To determine the strength of the interaction between CO2 molecules and the porous sorbents, the isosteric heat of adsorption was calculated by applying the Clausius–Clapeyron equation to the adsorption isotherms. The values found for these materials are in the 22–30 kJ mol−1 range, typical for a physisorption process and similar to other activated carbons.22,28 Those values are lower than for amine-functionalized materials, which are in the 40–90 kJ mol−1 range.29–31 This result suggests that the N functionalities present in these materials do not interact significantly with CO2 molecules, so that the adsorption of CO2 will be governed by the textural properties (vide infra). It should be mentioned that Simmons et al. showed for MOFs that, under typical swing adsorption processes, which are widely used in industrial processing, an isosteric heat of adsorption between 22 and 28 kJ mol−1 is optimal for single-component CO2 streams and between 26 and 31 kJ mol−1 for a 20% partial pressure CO2 flue gas.32 Taking this into account, these materials would be highly suitable for such CO2 capture systems. To further probe such statement, the regeneration of the sorbents was studied. To test the regeneration of the carbon sorbents, adsorption (CO2 at 1 bar) desorption (under He) cycles were performed at 25 °C in a TGA system. The results are shown in Fig. 3a for the CO2 sorbent with the highest uptake (AG-2-700). It can be seen that both CO2 adsorption and desorption processes are very fast, taking place in a span of 6 min. Furthermore, the adsorption-desorption cycles can be repeated several times and no noticeable changes are observed in the sorption kinetics nor CO2 uptake. This result is in agreement with the relatively weak interaction between CO2 and these sorbents.
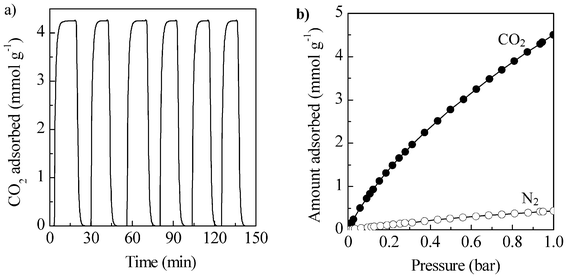 | ||
| Fig. 3 a) CO2 adsorption-desorption cycles for the sample AG-2-700 at 25 °C and 1 bar and b) CO2 and N2 isotherms at 25 °C for AG-2-700. | ||
| Sample | Amount of CO2 adsorbed, mmol g−1 (mg g−1) | Q st (kJ mol−1)a | ||
|---|---|---|---|---|
| 0 °C | 25 °C | 50 °C | ||
| a Isosteric heat of adsorption determined at a CO2 surface coverage of 0.7 mmol g−1. | ||||
| AG-1-700 | 6.7 (292) | 4.1 (178) | 2.5 (111) | 25 |
| AG-2-600 | 6.2 (273) | 4.2 (185) | 2.7 (118) | 30 |
| AG-2-650 | 7.0 (306) | 4.4 (192) | 2.8 (125) | 27 |
| AG-2-700 | 7.4 (325) | 4.5 (198) | 2.8 (123) | 25 |
| AG-4-600 | 6.2 (274) | 3.8 (167) | 2.5 (108) | 25 |
| AG-4-650 | 5.9 (260) | 3.5 (155) | 2.2 (97) | 24 |
| AG-4-700 | 6.1 (267) | 3.8 (166) | 2.5 (109) | 22 |
| Material | Sample code | Amount of CO2 adsorbed (mmol g−1) | Reference | ||
|---|---|---|---|---|---|
| 0 °C | 25 °C | 50 °C | |||
| N-doped algae-derived AC | AG-2-650 | 7.0 | 4.4 | 2.8 | This work |
| AG-2-700 | 7.4 | 4.5 | 2.8 | ||
| Sawdust-derived AC | AS-2-700 | 6.6 | 4.3 | 2.6 | 22 |
| AS-2-600 | 6.1 | 4.8 | 3.6 | ||
| TiC-CDC | micro-TiC 600 °C, H2 @ 600 °C | 7.1 | — | — | 1 |
| Petroleum pitch-derived AC | DO-88-M | 6.5 | 4.7 | 2.9 | 27 |
| VR-5-M | 8.6 | 4.2 | 2.3 | ||
| N-doped templated microporous carbons | CEM-750 | 6.9 | 4.4 | — | 2 |
| PAN fiber-derived AC | PAN-PK | — | 4.4 | — | 45 |
To probe the selectivity of the sorbents against the major component in flue gas, i.e. N2 (>70%), N2 isotherms were also measured at 25 °C for two sorbents, AG-2-650 and AG-2-700 (see Fig. 3b). In both cases, the amount of N2 adsorbed is much lower than the amount of CO2 (0.43 and 0.44 mmol g−1 for AG-2-650 and AG-2-700 respectively), which yields a good CO2/N2 equilibrium selectivity of around 10 in both cases.
The correlation of the textural and chemical characteristics of these materials with their CO2 adsorption capacity can provide useful information for the design of high-performance CO2 capture sorbents. In this respect, we found that no correlation exists between the apparent surface area nor the total pore volume and the CO2 uptake at 0 °C and 1 bar (likewise for 25 °C) (Fig. S4†). The same behavior is observed for activated carbons prepared from HTC sawdust, cellulose and starch (see Fig. S4†),22 activated templated carbons,33 microporous organic polymers4 and carbide-derived carbons.1 Presser et al. found, however, a linear correlation between the CO2 uptake at 0 °C and 1 bar and the volume of pores smaller than 0.8 nm determined applying the NLDFT method to the CO2 adsorption isotherm at 0 °C for CDCs and activated CDCs.1 The same trend is observed here when the CO2 uptake at 0 or 25 °C and 1 bar is depicted vs. the volume of narrow micropores (<0.7–0.8 nm) determined by applying the Dubinin-Radushkevitch equation to the CO2 isotherm (Vn (CO2)), as shown in Fig. 4a for 25 °C. Furthermore, the same trend is found for HTC sawdust-, cellulose- and starch-based porous carbons, materials that exhibited also a remarkable performance in CO2 capture,22 and for a variety of porous carbon materials, including N-doped carbons (with similar N-moieties to the materials here synthesized), that can be found in the literature (see Fig. 4a).2,34–38 These findings indicate that the adsorption of CO2 at sub-atmospheric conditions by porous materials is governed by the volume of narrow micropores and confirm the theoretical calculations that show that only pores of less than 1 nm can be effective for CO2 adsorption at atmospheric pressure.39,40 What is more, independently of the N or O contents of the samples, they all fit the same trend. This suggests a minor contribution of those functionalities to the CO2 uptake in comparison to the volume of narrow micropores, which is related to the non-polarity and weak polarizability of the CO2 molecule.41–43 In this respect, previous excellent performances that have been attributed to the N present in the carbon materials (excluding amine functionalization) would be actually due to their narrow microporosity development. Further evidence of the influence of the pore size in the CO2 capture capacity is observed when the CO2 uptake per volume of micropores (V0 (N2)) is represented vs. average pore width, L0 (see Fig. 4b). Thus, a clear decrease of the CO2 uptake per volume of micropores is registered with the enlargement of the average pore width for a variety of porous carbon materials. This behavior is similar to that observed for hydrogen adsorption.44 Overall, the results here presented indicate that the pore size is the key parameter that should be controlled in the design of porous sorbents for CO2 capture, trying to maximize the volume of narrow micropores. On the other hand, N-doping should be performed selectively to introduce amines, as less basic functionalities do not interact significantly with CO2 molecules.
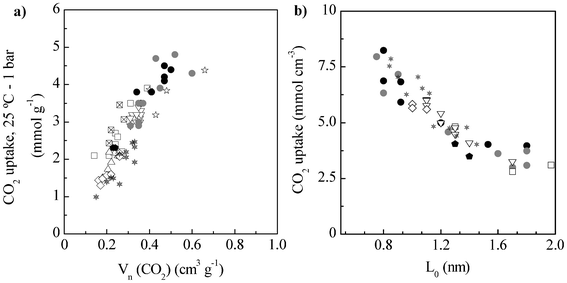 | ||
| Fig. 4 a) Correlation between the CO2 uptake at 25 °C and 1 bar and the volume of narrow micropores for a variety of porous carbon materials: • – algae-derived N-doped carbons (this work), – HTC-derived carbons (ref. 22), □ – polypyrrole-derived carbons (ref. 36), ∇ – activated templated carbons (ref. 33), – N-doped zeolite templated carbons (ref. 2), △ – N-doped activated carbons (ref. 34), ⋄ – ammoxidised carbons (ref. 35), – phenol-formaldehyde resin-derived activated carbons (ref. 37) – N-doped ordered mesoporous carbons (ref. 38) and – two commercial activated carbons measured in our lab (Super and Supra DLC-50 by Norit). b) Average pore width and CO2 uptake (at 0 °C and 1 bar) per total volume of micropores of the porous carbon materials. | ||




4. Conclusion
In conclusion, we have shown a promising approach for the synthesis of carbon-based sorbents for CO2 capture. It is based on a green and sustainable approach, i.e. the hydrothermal carbonization of biomass, which enables the transformation of microalgae into a highly microporous N-doped carbon material through a subsequent step of chemical activation. We have shown that the control of the activation conditions (temperature and amount of KOH) allows the synthesis of exclusively microporous biomass-based materials. These materials possess apparent surface areas in the ∼1300–2400 m2 g−1 range and pore volumes up to ∼1.2 cm3 g−1. They additionally exhibit N contents in the 1.1–4.7 wt% range, these heteroatoms being mainly present as pyridone-type structures. When tested as CO2 sorbents at sub-atmospheric conditions, they show large CO2 capture capacities, among the highest for porous materials, up to 7.4 mmol g−1 at 0 °C and 1 bar. This large CO2 capture capacity is exclusively due to their high volume of narrow micropores and not to the high surface areas or pore volumes, neither to the presence of heteroatoms. This result is a key point for the future design of high-performance CO2 sorbents. Additionally, those sorbents are easily and completely regenerated, without the energy-penalty associated to the use of amine-based sorbents or systems.Acknowledgements
The financial support for this research work provided by the Spanish MCyT (CQT2011-24776) is gratefully acknowledged. M.S. acknowledges the assistance of the Spanish MCINN for its award of a Ramón y Cajal contract.References
- V. Presser, J. McDonough, S. H. Yeon and Y. Gogotsi, Energy Environ. Sci., 2011, 4, 3059–3066 CAS.
- Y. Xia, R. Mokaya, G. S. Walker and Y. Zhu, Adv. Energy Mater., 2011, 1, 678–683 CrossRef CAS.
- D. D'Alessandro, B. Smit and J. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef CAS.
- R. Dawson, E. Stockel, J. R. Holst, D. J. Adams and A. I. Cooper, Energy Environ. Sci., 2011, 4, 4239–4245 CAS.
- N. Hedin, L. Chen and A. Laaksonen, Nanoscale, 2010, 2, 1819–1841 RSC.
- S. Choi, J. Drese and C. Jones, ChemSusChem, 2009, 2, 796–854 CrossRef CAS.
- M. Frac, S. Jezierska-Tys and J. Tys, Afr. J. Biotechnol., 2010, 9, 9227–9236 Search PubMed.
- Y. Chisti, Trends Biotechnol., 2008, 26, 126–131 CrossRef CAS.
- J. Singh and S. Gu, Renewable Sustainable Energy Rev., 2010, 14, 2596–2610 CrossRef CAS.
- A. Demirbas and M. Fatih Demirbas, Energy Convers. Manage., 2011, 52, 163–170 CrossRef.
- M. Terrones, P. M. Ajayan, F. Banhart, X. Blase, D. L. Carroll, J. C. Charlier, R. Czerw, B. Foley, N. Grobert, R. Kamalakaran, P. Kohler-Redlich, M. Rühle, T. Seeger and H. Terrones, Appl. Phys. A: Mater. Sci. Process., 2002, 74, 355–361 CrossRef CAS.
- Q. Yang, W. Xu, A. Tomita and T. Kyotani, Chem. Mater., 2005, 17, 2940–2945 CrossRef CAS.
- Carbon Materials for Catalysis, ed. P. Serp and J. L. Figueiredo, John Wiley & Sons, Inc., Hoboken, New Jersey, 2009 Search PubMed.
- ISO 9277:2010. Determination of the specific surface area of solids by gas adsorption – BET method. Second edition of ISO 9277, ISO, Geneva, 2012.
- F. Stoeckli and L. Ballerini, Fuel, 1991, 70, 557–559 CrossRef CAS.
- P. I. Ravikovitch and A. V. Neimark, Langmuir, 2006, 22, 11171–11179 CrossRef CAS.
- A. V. Neimark, Y. Lin, P. I. Ravikovitch and M. Thommes, Carbon, 2009, 47, 1617–1628 CrossRef CAS.
- C. Falco, M. Sevilla, R. J. White, R. Rothe and M. M. Titirici, ChemSusChem, 2012, 5, 1834–1840 CrossRef CAS.
- M. Sevilla, A. B. Fuertes and R. Mokaya, Energy Environ. Sci., 2011, 4, 1400–1410 CAS.
- M. Sevilla, N. Alam and R. Mokaya, J. Phys. Chem. C, 2010, 114, 11314–11319 CAS.
- M. Sevilla, R. Foulston and R. Mokaya, Energy Environ. Sci., 2010, 3, 223–227 CAS.
- M. Sevilla and A. B. Fuertes, Energy Environ. Sci., 2011, 4, 1765–1771 CAS.
- M. Sevilla, R. Mokaya and A. B. Fuertes, Energy Environ. Sci., 2011, 4, 1400–1410 CAS.
- L. Zhao, L. Z. Fan, M. Q. Zhou, H. Guan, S. Qiao, M. Antonietti and M. M. Titirici, Adv. Mater., 2010, 22, 5202–5206 CrossRef CAS.
- H. Wang, Q. Gao and J. Hu, Microporous Mesoporous Mater., 2010, 131, 89–96 CrossRef CAS.
- F. Kapteijn, J. A. Moulijn, S. Matzner and H. P. Boehm, Carbon, 1999, 37, 1143–1150 CrossRef CAS.
- A. Wahby, J. M. Ramos-Fernández, M. Martínez-Escandell, A. Sepúlveda-Escribano, J. Silvestre-Albero and F. Rodríguez-Reinoso, ChemSusChem, 2010, 3, 974–981 CrossRef CAS.
- K. T. Chue, J. N. Kim, Y. J. Yoo, S. H. Cho and R. T. Yang, Ind. Eng. Chem. Res., 1995, 34, 591–598 CrossRef CAS.
- B. Arstad, H. Fjellvag, K. O. Kongshaug, O. Swang and R. Blom, Adsorption, 2008, 14, 755–762 CrossRef CAS.
- Y. S. Bae and R. Q. Snurr, Angew. Chem., Int. Ed., 2011, 50, 11586–11596 CrossRef CAS.
- M. R. Mello, D. Phanon, G. Q. Silveira, P. L. Llewellyn and C. M. Ronconi, Microporous Mesoporous Mater., 2011, 143, 174–179 CrossRef CAS.
- J. M. Simmons, H. Wu, W. Zhou and T. Yildirim, Energy Environ. Sci., 2011, 4, 2177–2185 CAS.
- M. Sevilla and A. B. Fuertes, J. Colloid Interface Sci., 2012, 366, 147–154 CrossRef CAS.
- C. Pevida, M. G. Plaza, B. Arias, J. Fermoso, F. Rubiera and J. J. Pis, Appl. Surf. Sci., 2008, 254, 7165–7172 CrossRef CAS.
- M. G. Plaza, F. Rubiera, J. J. Pis and C. Pevida, Appl. Surf. Sci., 2010, 256, 6843–6849 CrossRef CAS.
- M. Sevilla, P. Valle-Vigón and A. B. Fuertes, Adv. Funct. Mater., 2011, 21, 2781–2787 CrossRef CAS.
- C. F. Martín, M. G. Plaza, S. García, J. J. Pis, F. Rubiera and C. Pevida, Fuel, 2011, 90, 2064–2072 CrossRef.
- L. Liu, Q. F. Deng, T. Y. Ma, X. Z. Lin, X. X. Hou, Y. P. Liu and Z. Y. Yuan, J. Mater. Chem., 2011, 21, 16001–16009 RSC.
- J. M. Martín-Martínez, R. Torregrosa-Maciá and M. C. Mittelmeijer-Hazeleger, Fuel, 1995, 74, 111–114 CrossRef.
- A. Vishnyakov, P. I. Ravikovitch and A. V. Neimark, Langmuir, 1999, 15, 8736–8742 CrossRef CAS.
- V. Yu. Korovin, V. V. Barkova and V. A. Platonov, Russ. J. Appl. Chem., 1994, 67, 237–240 Search PubMed.
- A. A. Énnan, V. S. Kovtun, V. V. Strelko and N. T. Kartel, Ukr. Khem. Zh., 1987, 53, 584–586 Search PubMed.
- I. A. Kuzin, A. I. Loskutov, V. F. Palfitov and L. A. Koémets, Zhurnal Prikladnoi Khimii, 1972, 45, 760–765 CAS.
- G. Yushin, R. Dash, J. Jagiello, J. G. Fischer and Y. Gogotsi, Adv. Funct. Mater., 2006, 16, 2288–2293 CrossRef CAS.
- W. Shen, S. Zhang, Y. He, J. Li and W. Fan, J. Mater. Chem., 2011, 21, 14036–14040 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: SEM images of N-doped hydrothermal carbon and N-doped activated carbon, N2 physisorption isotherms of the activated carbons, evolution of the apparent surface area and pore volume with the increase of the activation temperature for different KOH/HTC carbon weight ratios and correlation between the a) apparent surface area and b) total pore volume and the CO2 uptake (at 0 °C and 1 bar) of the porous carbon materials. See DOI: 10.1039/c2ra22552b |
| This journal is © The Royal Society of Chemistry 2012 |