Synthesis of a hydrogen producing nanocrystalline ZnFe2O4 visible light photocatalyst using a rapid microwave irradiation method†
Rekha
Dom
a,
R.
Subasri
a,
Neha Y.
Hebalkar
a,
A. Sadananda
Chary
b and
Pramod H.
Borse
*a
aInternational Advanced Research Centre for Powder Metallurgy and New Materials (ARC International), Balapur PO, Hyderabad, Andhra Pradesh, India 500005. E-mail: phborse@arci.res.in; Fax: 91-40-2444-2699; Tel: 91-40-2445-2426
bDepartment of Physics, University College of Science, Osmania University, Hyderabad, Andhra Pradesh, India 500007
First published on 23rd October 2012
Abstract
A rapid microwave solid-state synthesis method is systematically investigated to achieve a H2 producing visible light active spinel photocatalyst. ZnFe2O4 nanocrystallites were obtained by microwave irradiation of precursor compacts under optimized conditions. This investigation led to a uniform sized nanocrystalline photocatalyst that yielded a quantum-yield of H2 evolution ∼3.8 times higher than that of conventionally synthesized ZnFe2O4. The synthesis parameters – microwave power, synthesis temperature, and time, were found to control the physico-chemical properties viz phase formation kinetics, phase purity, crystallinity, specific surface area and photochemical efficiency, of the synthesized photocatalyst. The study reveals that the threshold microwave power of ≥3 kW was necessary to obtain a spinel phase structure, while lower power (<3 kW) could not induce the crystallization even after prolonged low-power irradiation of 180 min. At the threshold power, a minimum of 10 min. synthesis time was enough to obtain uniform sized nanocrystallites, indicating that the synthesis method is ∼24 times faster than the solid state reaction method, which needs nearly 4 h. The particle morphology evolution with irradiation time from 10–150 min. exhibited de-crystallization phenomena. Longer irradiation displayed a morphological crystallization probably induced due to the simultaneous area and volumetric heating effect. The possible “formation mechanism” of these uniform nanocrystallites has been presented here for qualitative understanding. Thus synthesized photocatalysts generated hydrogen from a water–methanol mixture even without the co-catalyst loading. The ferrite photocatalyst was found to decolorize methylene blue dye with a maximum decay constant of 0.232 h−1, thereby demonstrating its capability in the pollutant decomposition applications, all under visible light photons.
1. Introduction
For a large scale application of hydrogen-energy-generating or pollutant-degrading visible-light-photocatalysts, it is highly desirable to develop an easy, quick and green-synthesis production method. In addition to low-energy photon absorptivity and well suited band energetics of the material system;1–5 a synergy between the crystallinity and surface area of the catalytic particle plays a crucial role in obtaining an efficient particulate photocatalyst.1,6,7 Microwave synthesis is one such synthesis method, which yields high crystallinity nanocrystallites in a short processing time.8 To date, microwave radiation (0.3 GHz–300 GHz) has found applications in a number of fields, ranging from microwave communication to medical diagnostics, treatment and radiation induced material processing techniques.8–10 These waves undergo absorption, transmission or reflection when they interact with matter viz, metal, ceramics.11–13 Its ability for full scale ceramic product processing is its peculiar advantage. This is because these electromagnetic waves directly interact with the full volume of material at a molecular level via di-polar interactions, thus penetrating through the material to deliver the energy, and generate cubical heating of the material. Consequently, it is possible to achieve a rapid and uniform heating of materials.14 Dielectric materials, such as ferrites, are well suited for microwave sintering, as well as possessing opto-electronic properties desirable for visible light photocatalysis.3,4–15,16 Thus ferrites are chosen for the present study. Recently, there have been several reports on the synthesis of ZnFe2O4 (ZFO) particles by using various techniques viz solid state reaction (SSR), sol–gel, co-precipitation hydrothermal, hydrothermal-microwave method, sonochemical and pyrolysis methods.17–21 However, these methods need a longer synthesis time and yield low-crystallinity poly-dispersed particles. In addition, they involve multi-step procedures, mostly involving post-calcination treatment to achieve the desired crystal phase. Surprisingly, there are very few reports22–24 on the microwave synthesis of ternary ferrites. Motivated by our recent work on the use of microwaves for producing high crystallinity nano-sized particles, we took this work to understand the tunability of material properties with respect to various experimental parameters, so as to achieve an efficient photocatalyst for solar hydrogen generation and pollutant degradation application. It is believed that such study of parametric dependence of the material properties is highly important to tune and achieve desirable material properties. It is important to note that high crystallinity particles are highly desirable and useful in photocatalysis research, as is evident here.There is tremendous interest to develop an eco-friendly and low band gap material that can be utilized for photocatalytic hydrogen generation under visible light. Accordingly, ZFO was found to be one of the best suited oxide as a photocatalyst. It is an n-type semiconductor that exhibits a relatively a low band gap (∼1.9 eV), and suitable band energetics,6,7 thus making it a useful material for solar hydrogen production from water splitting. In recent reports6,7 it was showed that a ZFO photocatalyst prepared by SSR and a polymer complex method can photo-decompose phenol, as well as photo-reduce water under visible light photons. Similarly, Cao showed that ZFO prepared by a microwave-hydrothermal method was found to be useful in the photodecomposition of phenol.25 The nanostructured floriated ZFO nanorods displayed excellent photocatalytic hydrogen producing capacity.26 The nano-composite27 constituting ZFO–TiO2 made by hydrothermal treatment was found to be useful for the photodegradation of Rhodamine. An enhanced photocatalytic hydrogen evolution was observed by Boumaza. et al. over a ZFO–SrTiO3 hetero-system photocatalyst.28 The nano-composite of ZFO/MWCNT (multi walled carbon nanotube) was found to be 99% efficient for photodegradation of methylene blue29 under visible light irradiation. Very recently Fu et al. demonstrated most efficient photodegradation of methylene blue with a ZFO–graphene photocatalyst.30 Similar results were also demonstrated by Kong et al. by using ZFO–BiOBr hetero-junctions.31 These studies display the great potential of ZFO as a solar active photocatalyst.
In the present study, we have systematically attempted a careful parametric optimization of microwave sintering to realize the high crystallinity pure phase ZFO photocatalyst, all without any need of post-calcination steps. Thus to achieve the desired ZFO phase, the prominent experimental synthesis parameters, viz microwave power, synthesis time and temperature, etc. were systematically varied to investigate their influence on the physico-chemical properties viz. phase purity, crystallinity, specific surface area and photocatalytic behavior; of the formed ferrite nanocrystals. The synthesized particles were characterized in-depth for their structure, morphology and physico-chemical properties using necessary characterization techniques, and investigated to demonstrate their solar hydrogen producing capacity. The study clearly reveals that the magnitude of microwave power is an important factor responsible for the pure phase formation of nanocrystalline ZFO.
2. Experimental
2.1. Microwave synthesis of nanocrystalline ZnFe2O4
Desired spinel phase photocatalysts were synthesized by a systematic optimization of the parameters involved in this microwave synthesis methodology. In a typical synthesis, in each case, ZnO (99%, Merck) and Fe2O3 (98.5%, Loba) powders were mixed and ground in methanol, and further used to form the powder compacts. The powder compacts were further subjected to an electromagnetic irradiation using a microwave furnace (PLC controlled 6.4 kW, model MHTD supplied by Linn High Therm GmbH). The microwave power (1–6 kW), reaction temperature (773–1173 K) and the sintering time (10–150 min.) was varied to achieve the desired phase in the photocatalyst. The irradiated powder compacts were used for further characterization. In order to test the formation of ZFO at very low power, a 1 kW microwave oven (conventional kitchen oven) was first utilized to perform the experiments. However the results were not encouraging, as the microwave power was not sufficient to induce the spinel ZFO phase formation. Thus the remaining experiments were carried out in the laboratory microwave furnace. In order to carry out a detailed study, preliminary investigation of influence of several parameters were carried out as described in the following sections, to achieve a pure spinel ZFO phase. Further, in order to compare the properties of microwave sintered samples, a standard reference sample was synthesized by the conventional SSR method.6 Briefly, the reference samples were synthesized by mixing the stoichiometric mixture of Zn and Fe oxides and further pelletized. Thus produced samples were calcined at different temperatures of 1073–1273 K for 4–10 h in a static air furnace. These were further used for characterization. The powders were used for the photocatalytic hydrogen evolution and pollutant degradation experiments.2.2. Characterization
The samples were comprehensively characterized before being subjected to the photochemical experiments. Structural phase confirmation being the most important objective, X-ray diffraction (XRD) studies of all the samples were carried out. An advanced XRD equipment (Bruker AXS D8) that is equipped with a Lynx eye detector and Cu-Kα (1.542 Å) radiation was used to ascertain the sample phase compositions, within the detection limit characteristics of this technique. The accelerating potential and the filament current were fixed to 40 kV and 40 mA, respectively. The XRD scan was recorded in the range of 20–80° 2θ angles with a step size of 0.01°. The specific surface area of the powder samples was estimated from the Brunauer–Emmett–Teller (BET) method by nitrogen adsorption at 77 K, using a BET model Micrometrics, ASAP2020 V3.00 H analyzer. The samples were degassed at 343 K prior to the respective specific surface area measurements. The optical properties were studied by UV-Visible Diffuse reflectance spectroscopy (DRS) using a Perkin-Elmer Lambda-750 spectrometer in the wavelength range 200–800 nm. The raw data was further analyzed using Tauc analysis. The qualitative investigations of sample morphology were carried out using field-emission scanning electron microscopy (Hitachi S4300SE/N) with the instrument operated at 20 kV, and equipped with an energy-dispersive X-ray analyzer (EDAX USA, PV7747/36ME). The sample microstructure was further characterized by high-resolution TEM (HRTEM) using a Technai 210 TEM working at 200 kV, and was equipped with an energy dispersive X-ray spectroscopy (EDS) system. The elemental composition and respective electronic state was investigated by X-ray photoelectron spectroscopy (XPS), XPS Model Omicron with an Al-Kα (1486.8 eV) radiation source. The binding energy was calibrated using internal calibration of C1s with reference energy at 285.5 eV. Raman studies were carried out using a Jobin Yuan, HR800 micro Raman model at the 514.5 nm Ar+ laser line.2.3. Photocatalytic characterization for methylene blue decomposition and solar hydrogen generation
The photocatalytic behavior of the as-synthesized samples was studied by carrying out pollutant photodegradation and photocatalytic hydrogen generation studies. Methylene blue (MB) was chosen as an organic pollutant for the photocatalytic degradation studies. The MB degradation was carried-out in a special, indigenously designed and fabricated vertical illumination reactor (100 ml) containing a top quartz window, that was placed under solar simulator (300 W, Model-91160-New Port Oriel instruments) equipped with an AM1.5G filter. In each case, an aqueous MB solution (1 mg per 100 ml) containing photocatalyst (60 mg) was used. Prior to the photocatalytic reaction, the mixture was stirred in the dark for one hour, to establish the adsorbate-substrate equilibrium. The system was maintained at room temperature during the experimentation. An aliquot of 5 ml was extracted from the main liquor and analysed for MB concentration by recording the absorption spectra using a UV-Visible spectrophotometer. The supernatant obtained after centrifugation was used for this purpose. The results were correlated with the photo-catalytic degradation of MB to evaluate performance by estimating the de-coloration efficiency, η = [C − C0]/C0, where C0 and C are the respective MB concentrations before and after the photocatalytic reactions.The photocatalytic performance of the samples prepared by microwave synthesis were compared, and evaluated compared to that of ZFO prepared by the SSR method. It is important to state that MB de-coloration is a generally accepted way to investigate the photocatalytic capability of a photocatalyst. However, one must note that the redox-reaction would differ with degradant chemical. In order to validate the capability for the photo-splitting of water of ZFO nanoparticles, we continued the investigation to study the photo-reduction of a water–methanol mixture.
Accordingly, the photocatalytic hydrogen evolution (over ZFO) was studied under visible light irradiation (λ ≥ 420 nm) and solar simulator (AM1.5G filter). The reactions were carried out in specially designed and fabricated horizontal illumination and vertical illumination closed gas reactors, which were used under a Hg-arc lamp and a solar simulator, respectively. The Hg-arc lamp was connected to a collimator and infrared (IR) water filter to avoid heating of the reaction mixture. A monochromatic filter was used to select the visible light (λ = 420 nm) for carrying out the reaction. For this 50 mg of the photocatalyst was taken in a pyrex cell containing a water–methanol mixture and the solution was purged with N2 prior to irradiation. The rate of H2 evolution was detected using a gas chromatograph (Shimadzu 2010AFT) equipped with a thermal conductivity detector. For hydrogen evolution studies, typically light was made “ON” for a certain duration of time and hydrogen gas evolved was estimated. Under Light “OFF” there was no evolution of gas. The quantum yield (QY) for hydrogen evolution using different samples were estimated using the procedure described earlier,1 using the following relation32
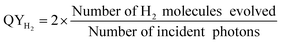 | (1) |
3. Results and discussion
3.1. Structural investigation of nanocrystalline ZnFe2O4
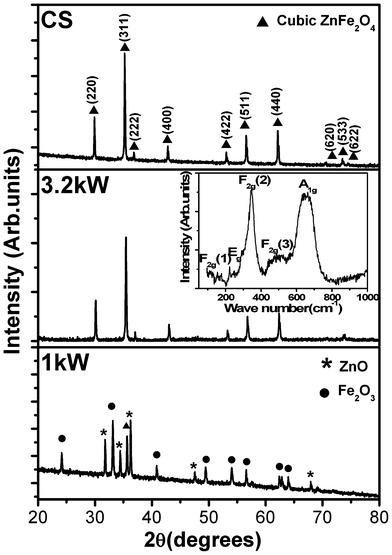 | ||
| Fig. 1 X-ray diffraction spectra of nanocrystalline ZnFe2O4 synthesized at a microwave power of 1 kW, 3.2 kW, and conventional sintering (CS). The inset shows the Raman spectrum of the compact obtained after the sintering at a microwave power of 3.2 kW. | ||
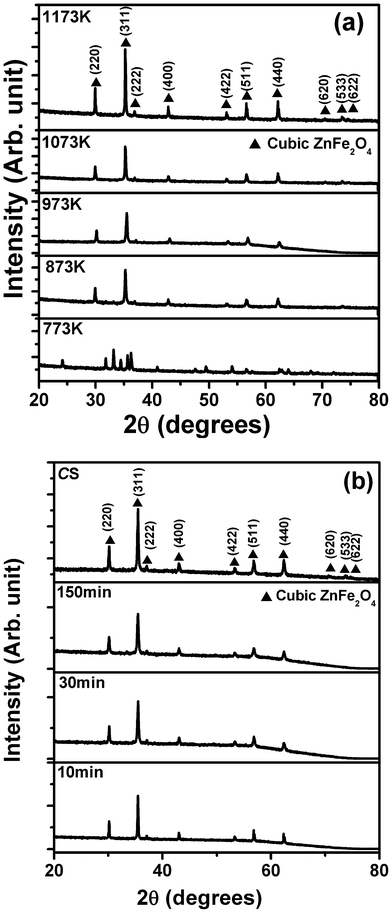 | ||
| Fig. 2 X-ray diffraction spectra of ZnFe2O4 samples synthesized by microwave sintering at, (a) different temperatures; and (b) for different times. The spectrum corresponding to conventional sintering is also shown as a reference. | ||
Fig. 2(b) shows the XRD spectra of the μS samples (T ∼ 973 K) irradiated for various time durations of 10, 30, 150 min, and the CS sample. Interestingly, all samples exhibited the spinel phase, and further confirm that a single phase can be acquired even in 10 min of microwave irradiation under the given conditions. It is noteworthy to mention that, in the case of microwave sintering, a shorter processing time is required to achieve the desired phase (Fig. S.I.2, ESI†).
Further, the effect of sintering temperature can be clearly understood from Fig. 3. The crystallite size and the ZFO (311) peak intensity display a linear trend in their variation with the sintering temperature, indicating its similar behaviour with the thermally induced crystallization in the SSR technique.
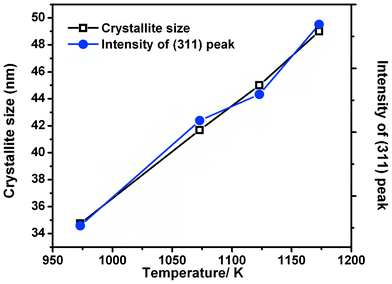 | ||
| Fig. 3 Crystallite size and intensity of (311) peak of ZnFe2O4 samples sintered at different temperatures during the microwave irradiation. | ||
For this, the crystallite size was estimated using the full width at the half maximum of the ZFO main peak (311), from the following Debye–Scherrer formula.35
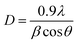 | (2) |
Where λ is the wavelength of the incident X-rays (0.154 nm), θ is the Bragg angle and β is the full width at the half maximum of the main peak (311).
Specifically, the trend in the variation in the peak intensity and consequent crystallite size with temperature shown in Fig. 3 implies that there is temperature dependent growth of the particle, along with an improvement in the crystallinity.
3.2. Morphological study of ZnFe2O4 nanoparticles
Fig. 4 shows the time dependent morphology changes in the ZFO synthesized by the μS method, and compared with those synthesized by the CS method. A qualitative analysis clearly demonstrates that the μS method yields smaller, distinct, and nearly uniform sized nanocrystallites than those prepared by the CS method. The CS sample displays the necked particle agglomerate. Further analysis indicates that the nano-crystal shape remained uniform when the irradiation time was varied from 10 min to 30 min; however, later the particles seem to reform the shape and acquire much smaller dimensions, as evident from the 150 min sample (see Fig. 4). Such de-crystallization phenomena is known to occur during the microwave phase sintering process.8,14 Analogous observations of reduction in crystallite size with microwave irradiation time were also confirmed from the XRD studies (See Table 1 and Fig. S.I.2, ESI†). The particle size variation is also in correlation with the variation in the specific surface area, as given in Table 1. | ||
| Fig. 4 FESEM images of nanocrystalline ZnFe2O4 synthesized by the CS method and the μS method for various irradiation times. | ||
| Sample | Crystallite size (nm) | Surface area (m2 g−1) | Band gap (eV) | Rate constant (k × 10−1/h) | Rate of hydrogen evolved (μmol (h g.cat)−1) | Quantum yield (%) | |
|---|---|---|---|---|---|---|---|
| Direct | Indirect | ||||||
| μS 10 min | 47 | 2.3 | 1.93 | 1.83 | 1.390 | 99.2 | 0.14 |
| μS 30 min | 35 | 4.6 | 1.93 | 1.83 | 2.320 | 133.5 | 0.19 |
| μS 150 min | 29 | 5.6 | 1.93 | 1.83 | 0.005 | 92.2 | 0.13 |
| CS | 53 | 2.2 | 1.9 | 1.81 | 2.150 | 31.7 | 0.05 |
In order to get further insight of the nature of the crystallites, TEM and HRTEM studies were done. Fig. 5 shows the TEM and HRTEM images of the ZFO nanocrystallites synthesized by the μS method. The d-spacing of 0.254 nm was found to match with the reflections from the (311) peak of ZFO. Thus the TEM study shows that high crystallinity crystallites have been achieved, as deduced from the XRD studies. It is desirable that a photocatalyst exhibits high crystallinity to render the catalytic site during the photocatalytic reaction. The above section clearly demonstrates the formation of high crystallinity, uniform particles, desirable for a photocatalyst.
 | ||
| Fig. 5 TEM and HRTEM images of the ZnFe2O4 nanocrystallites synthesized by μS method. | ||
3.3. Optical investigation of spinel ZnFe2O4
The optical properties play a vital role in determining the photocatalytic activity of any semiconducting material system. Fig. 6 shows the DRS spectra of the μS samples, which were obtained at different irradiation times viz 10 min, 30 min, and 150 min. The spectrum for the CS sample is also included for comparison. Interestingly, the μS samples showed higher absorbance w.r.t. CS sample, implying they can efficiently absorb visible light photons (of λ < 550 nm). A typical ZFO absorption curve is observed for all μS samples. which displayed a rather steep absorption unlike the CS sample, indicating that such an absorption can be attributed to an intrinsic band transition.36–38 It is known that, in spinel ZnFe2O4, the band structure is defined by the O2p valence band and the Fe3d conduction band. Thus, the ZnFe2O4 absorption can be attributed to the photo-excited electron transition of O2p → Fe3d.34 The sharp absorption on-set of the μS samples compared to the CS sample indicate that the μS sample possesses less structural defects, which is in accordance with the structural characterization discussed in the above section. Surprisingly, there were no significant differences in absorption spectra and band gaps of μS samples that were subjected to prolonged microwave irradiation.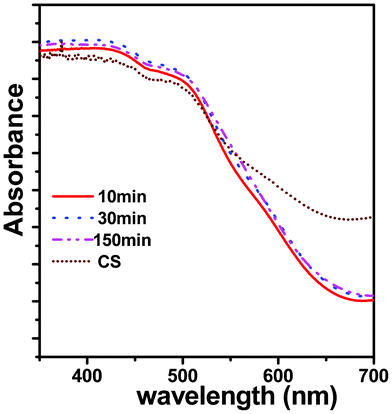 | ||
| Fig. 6 Diffuse reflectance spectra of nanocrystalline ZnFe2O4 synthesized by the μS method for various irradiation times (10–150 min). | ||
The optical band gap (Eg) was estimated from the absorption coefficient (α) using the Tauc relation39 given by
| αhν = A(hν − Eg)q | (3) |
![[/]](https://www.rsc.org/images/entities/char_e0ee.gif) for forbidden direct electronic transitions. These q values depend on the nature of the electronic transition within the band gap of semiconductor (q = ½ direct and 2 indirect here). Fig. 7 shows Tauc plots of the μS sample (30 min) that was used for the estimation of direct and indirect band gap of the ZFO nanocrystallites. The estimated band gaps were found to be 1.93 eV (direct) and 1.83 eV (indirect), as shown in Table 1, these values are similar to the bulk counterpart of ZFO (CS sample). As observed in the DRS spectra, all the μS samples yielded the same band gap values. The lower band gap values are useful for absorbing visible light photons and thus are useful in visible light photocatalysis.
for forbidden direct electronic transitions. These q values depend on the nature of the electronic transition within the band gap of semiconductor (q = ½ direct and 2 indirect here). Fig. 7 shows Tauc plots of the μS sample (30 min) that was used for the estimation of direct and indirect band gap of the ZFO nanocrystallites. The estimated band gaps were found to be 1.93 eV (direct) and 1.83 eV (indirect), as shown in Table 1, these values are similar to the bulk counterpart of ZFO (CS sample). As observed in the DRS spectra, all the μS samples yielded the same band gap values. The lower band gap values are useful for absorbing visible light photons and thus are useful in visible light photocatalysis.
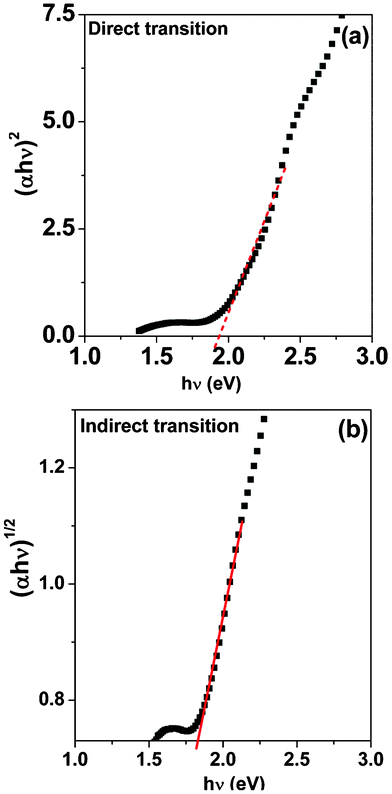 | ||
| Fig. 7 Tauc plots of μS 30 min. showing the (a) direct; and (b) indirect transition in the ZnFe2O4 band gap. | ||
3.4. X-ray photoelectron spectroscopy study of nanocrystalline ZnFe2O4
Fig. 8 displays the XPS analysis of the 30 min μS sample. The survey spectrum (Fig. 8(a)) clearly reveals the existence of Zn, Fe and O expected for ZnFe2O4 system, along with the adventitious C. Their core level spectra were further analyzed to find out the surface chemical composition and their respective oxidation states.20 The Zn doublet-splitting in Fig. 8(b) at 1021.5 eV (2p3/2) and 1044.7 eV (2p1/2) can be attributed to the +2 oxidation state of Zn. Fig. 8(c) indicates the Fe 2p peaks at 711.5 eV and 725.4 eV with a satellite at 718.6 eV, confirming that Fe exists in the +3 oxidation state. It is noteworthy that de-convolution studies indicate the possibility of the presence of a Fe2O3 surface impurity layer, which was not detected in the XRD studies. The O1s peak observed in Fig. 8(d) is correlated to oxygen existing in the ZnFe2O4 lattice.20 Analysis reveals that the ZFO composition contain a stoichiometric Zn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe 1:2 ratio as expected for a ZnFe2O4 system, and further indicates the oxidation states of Zn and Fe as +2 and +3, respectively.
:Fe 1:2 ratio as expected for a ZnFe2O4 system, and further indicates the oxidation states of Zn and Fe as +2 and +3, respectively.
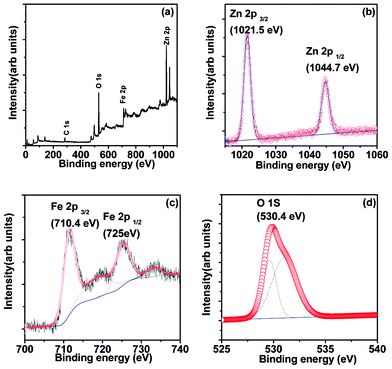 | ||
| Fig. 8 XPS of ZnFe2O4 prepared by the μS method showing, (a) survey spectra; and the core level spectrum for (b) Zn 2p; (c) Fe 2p; and (d) O 1s states. | ||
3.5. Study of photocatalytic properties of nanocrystalline ZnFe2O4
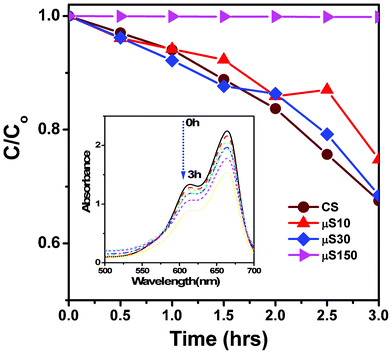 | ||
| Fig. 9 Photo-degradation of methylene blue as a function of illumination time (h), over various photocatalysts. The inset shows the optical absorption spectrum of methylene blue with the 30 min μS sample recorded at various illumination times. | ||
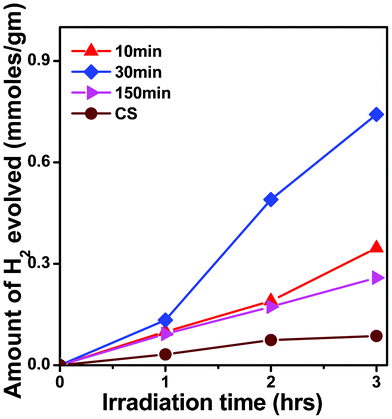 | ||
| Fig. 10 Time dependent photocatalytic H2 evolution from different photocatalysts under visible light photons (λ ∼ 420 nm); Photocatalyst: 100 mg ZFO in 70 ml H2O + 30 ml methanol (hole scavenger), with 500 W Hg-arc lamp. | ||
It is important to mention that metal co-catalysts are usually loaded on the semiconductor oxide photocatalyst to enhance H2 production by reducing the over-potential losses.1 Especially, in our case, it was not necessary to load co-catalyst over the photocatalyst surface of the μS sample. This implies that this simple method of photocatalyst formation has high potential for photocatalyst synthesis in industries. As far as Pt loaded ZFO7 was concerned, it was found that Pt-loaded ZFO can further enhance the H2 evolution, which can be attributed to the fact that Pt renders an easy passage to electron transfer from the photocatalyst surface to the solution. The metal/semiconductor interface acts as a Schottky barrier junction that can suppress the photogenerated electron–hole recombination. We presume that in the absence of Pt loading, the surface states available in the μS sample would be possibly playing the role of suppression of electron–hole recombination, which is not so in the case of the CS sample. The detailed investigation is underway and will be published elsewhere.
In brief, the μS photocatalysts were found to be better photocatalysts than those made by the conventional method. Table 1 clearly demonstrates that these low band-gap nanocrystalline photocatalysts behave in similar manner for (i) MB photodegradation, as well as for (ii) photo-reduction of water to yield H2. Both the values, the MB decay constant (k) and amount of H2 gas evolution were found to be enhanced for these μS sample photocatalysts. The excellent photocatalytic activity necessitates the need to improve the understanding of photocatalysts formed using the μS method, which are analyzed in the following section.
3.6. Mechanism and performance of photocatalyst formed using μSversusCS method
It has been found that the ZFO photocatalyst formed by the μS method yields improved photocatalytic properties over those synthesized by conventional methodology. In order to understand improvement in the photochemical properties of the μS photocatalyst, in the first instance it is important to understand the mechanism underlying the formation of photocatalysts, and secondly, to understand the working of these photocatalysts.Accordingly, as per observations made by the physico-chemical characterization of photocatalysts, we present a model (Fig. 11(a)) to explain the μS photocatalyst formation. In the microwave sintering method, it is known that the microwave energy is directly delivered to the material through molecular interaction, leading to instant volumetric heating of the material.11,24,37,40,41 This is in total contrast to the conventional solid state reaction method, where the thermal energy is delivered to the geometrical surface of the material via radiant or convection heating, which is in turn transferred to the bulk of the material via conduction.41 ZFO being a good dielectric material, it becomes a potential candidate for microwave processing. This property enables one to achieve rapid and uniform heating of thick material.24 Based on the physico-chemical investigations on the microwave and conventional particles, a schematic is proposed indicating the main difference in the formation of ZFO in μS and CS methods.
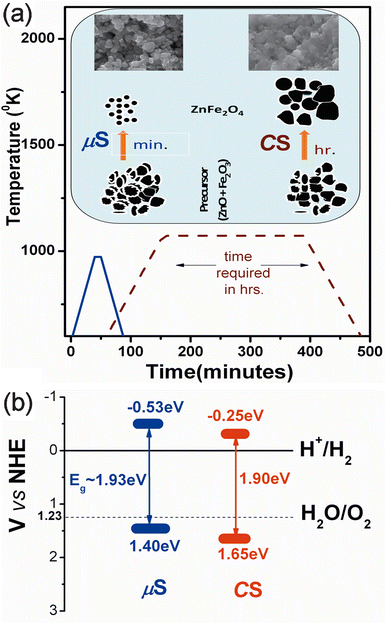 | ||
| Fig. 11 (a) Comparison of processing time (min) versus processing T/K for μS (solid line) and CS (dotted line) methods along with a schematic showing the possible mechanism for particle formation in the μS and CS methods. The SEM images show the morphology of ZFO formed during the respective processing; (b) a schematic band diagram for μS and CS photocatalysts as deduced from the optical and flat-band potential measurements. | ||
Fig. 11(a) displays the graph of processing time (irradiation in the μS and calcination in the CS method) versus calcination temperature required for formation of ZFO. The inset clearly displays the mechanism of particle formation in two cases. In the μS method, the instantaneous thermal shock to the powder compact induces a shape and size reformation, thus yielding comparatively smaller entities, as observed in the morphological as well as structural studies in the above sections. They agglomerate only after prolonged microwave irradiation, as shown in the FESEM micrographs (see Fig. 4 150 min sample). In contrast, in the CS method, the continuous thermal treatment of the powder compact facilitates the diffusion related inter, intra grain boundary diffusion, and related processes, leading to the formation of larger agglomerates. Comparison of the processing times in the two methods shows that the diffusion related phenomena (in CS) can be arrested to great extent in the microwave sintering step, which in turn retains or reduces the crystallite sizes. This is supported by the structural and morphological observations carried out in the respective sections. This fact is schematically explained in the inset of Fig. 11, which indicates the retention of nano-sized particles in the μS method in contrast to the CS method. This demonstrates that the μS method is a quick way to synthesize nanocrystalline ZFO as compared to the CS method.
The mechanism underlying the photocatalytic behaviour of the photocatalyst thus becomes important, to explain the enhanced visible light photocatalytic hydrogen generation from the μS sample. The optical band gap, band energetics and electrochemical properties are the important factors to understand the photochemical behaviour of a photocatalyst. An optical characterization of the ZFO clearly confirms that the band-gap lies in the visible wavelength range, as given in Table 1. The experimental flat-band potential measurements (Fig. S.I.4, ESI†) of photocatalysts and the theoretical electronic structure calculations (for bulk ZFO, Fig. S.I.5, ESI†) indicate that the valence band and conduction band of the ZFO system straddles the water redox-levels, as shown in Fig. 11(b). Interestingly the μS sample shows the conduction band edge at a more negative values than the CS sample, making it a potential photocatalyst for hydrogen evolution. The H2 evolution data for these photocatalyst is given in Table 1. Further it is evident (Fig. S.I.6 and Table S,I.-1, ESI†) from the electro-chemical impedance spectroscopy (EIS) that the μS sample shows larger donor density (ND ∼ 2.31 × 1021 m−3) in contrast to 6.74 × 1018 m−3 for the CS sample), which favours efficient hydrogen evolution during photocatalytic reactions. Thus the above analysis clearly validates the efficient behaviour of μS sample over the CS sample, thus demonstrating that microwave methodology is one of the best industrially viable options for the quicker synthesis of water splitting photocatalysts.
Conclusions
A rapid microwave synthesis method is explored to achieve a nanocrystalline photocatalyst that yielded a quantum-yield of H2 evolution ∼3.8 times higher than that of conventionally synthesized ZnFe2O4. During the systematic variation in the synthesis parameters, the microwave power, synthesis temperature, and time, altered the physico-chemical properties viz phase formation kinetics, phase purity, crystallinity and specific surface area of the synthesized photocatalyst. Highly crystalline, uniformly sized spinel ZnFe2O4 nanocrystallites were obtained by microwave irradiation of precursor compacts under optimized conditions. The study demonstrates that under given conditions, the microwave power of ≥3 kW is necessary to obtain a spinel phase structure, while a lower power (<3 kW) does not induce the crystallization even after prolonged low-power irradiation. A synthesis time of a minimum 10 min yields uniform sized nanocrystallites, demonstrating that this synthesis method is ∼24 times faster than the solid state reaction method, which needs nearly 4 h. The particle morphology evolution with irradiation time from 10–150 min. exhibits de-crystallization phenomena. The possible “formation mechanism” of these uniform nanocrystallites has been presented here. Thus-synthesized nanocrystallites photocatalysts efficiently generate hydrogen from a water–methanol mixture as compared to a conventional counterpart. They decolorize methylene blue dye, thereby demonstrating their capability in pollutant decomposition applications, all under visible light.Acknowledgements
The authors acknowledge Dr K. Radha and Dr B. V. Sarada for their help in BET and Raman measurements. Mr. K. Ramesh Reddy, and L. Venkatesh, are also acknowledged for their help in characterization.References
- H. G. Kim, P. H. Borse, W. Y. Choi and J. S. Lee, Angew. Chem., Int. Ed., 2005, 44, 4585–4589 CrossRef CAS.
- E. Casbeer, V. K. Sharma and X. Z. Li, Sep. Purif. Technol., 2012, 87, 1–14 CrossRef CAS.
- H. G. Kim, P. H. Borse, J. S. Jang, E. D. Jeong, O.-S. Jung, Y. J. Suh and J. S. Lee, Chem. Commun., 2009,(39), 5889–5891 RSC.
- P. A. Mangrulkar, V. Polshettiwar, N. K. Labhsetwar, R. S. Varma and S. S. Rayalu, Nanoscale, 2012, 4, 5202–5891 RSC.
- K. N. Harish, H. S. B. Naik, P. N. Prasanth Kumar and R. Viswanath, Catal. Sci. Technol., 2012, 2, 1033–1039 CAS.
- J. S. Jang, S. J. Hong, J. S. Lee, P. H. Borse, O. K. S. Jung, T. E. Hong, E. D. Jeong, M. S. Won and H. G. Kim, J. Korean Phys. Soc., 2009, 54, 204–208 CrossRef CAS.
- P. H. Borse, J. S. Jang, S. J. Hong, J. S. Lee, J. H. Jung, T. E. Hong, C. W. Ahn, E. D. Jeong, K. S. Hong, J. H. Yoon and H. G. Kim, J. Korean Phys. Soc., 2009, 55, 1472–1477 CrossRef CAS.
- D. Louzguine, G. Luzgin, Q. Xie, S. Li, A. Inoue, N. Yoshikawa, K. Mashiko, S. Taniguchi and M. Sato, J. Alloys Compd., 2009, 483, 78–81 CrossRef.
- M. Sorescu, L. Diamandescu, P. D. Ramesh and R. Roy, Mater. Chem. Phys., 2007, 101, 410–414 CrossRef CAS.
- S. Das, A. K. Mukhopadhyay, S. Satta and D. Basu, Bull. Mater. Sci., 2009, 31, 943–956 CrossRef.
- K. Saitou, Scr. Mater., 2006, 54, 875–879 CrossRef CAS.
- W. H. Sutton, Ceram. Bull., 1989, 68, 376–386 CAS.
- E. T. Thostenson and T.-W. Chou, Composites, Part A, 1999, 30, 1055–1071 CrossRef.
- R. Roy, R. Peelamedu, C. Grimes, J. Cheng and D. Agrawal, J. Mater. Res., 2002, 17, 3008–3011 CrossRef CAS.
- E. D. Jeong, P. H. Borse, J. S. Jang, J. S. Lee, C. R. Cho, J. S. Bae, S. Park, O. S. Jung, S. M. Ryu and H. G. Kim, J. Nanosci. Nanotechnol., 2008, 8, 3568–3573 Search PubMed.
- P. Xiong, Q. Chen, M. G. He, X. Sun and X. Wang, J. Mater. Chem., 2012, 22, 17485–5891 RSC.
- D. Makovec and M. Drofenic, J. Nanopart. Res., 2008, 10, 131–141 CrossRef CAS.
- J. M. Yang and K. L. Yang, J. Nanopart. Res., 2009, 11, 1739–1750 CrossRef CAS.
- A. Kundu, S. Anand and H. C. Verma, Powder Technol., 2003, 132, 131–136 CrossRef CAS.
- M. Wang, Z. Ali and L. Zhang, J. Phys. Chem. C, 2008, 112, 13163–13170 CAS.
- M. Sivakumar, T. Takami, H. Ikuta, A. Towata, K. Yasui, T. Tuziuty, T. Kozuka, D. Bhattacharya and Y. Iida, J. Phys. Chem. B, 2006, 110, 15234–15243 CrossRef CAS.
- P. Yadoji, R. Peelamedu, D. Agrawal and R. Roy, Mater. Sci. Eng., B, 2003, 98, 269–278 CrossRef.
- Y. P. Fu, Mater. Res. Bull., 2006, 41, 809–816 CrossRef CAS.
- M. Sertkol, Y. Koseoglu, A. Baykal, H. Kavas, A. Bozkurt and M. S. Toprak, J. Alloys Compd., 2009, 486, 325–329 CrossRef CAS.
- S. W. Cao, J. Hazard. Mater., 2009, 171, 431–435 CrossRef CAS.
- L. V. Hongjin, L. Ma, P. Zeng, D. Ke and T. Peng, J. Mater. Chem., 2010, 20, 3665–3672 RSC.
- G. Y. Zhang, Y. Q. Sun, D. Z. Gao and Y. Y. Xu, Mater. Res. Bull., 2010, 45, 755–760 CrossRef CAS.
- S. Boumaza, A. Boudjemaa, A. Bouguelia, R. Bouarab and M. Trari, Appl. Energy, 2010, 87, 2230–2236 CrossRef CAS.
- C.-H. Chen, Y. H. Liang and W.-D. Zhang, J. Alloys Compd., 2010, 501, 168–172 CrossRef CAS.
- Y. Fu and X. Wang, Ind. Eng. Chem. Res., 2011, 50, 7210–7218 CrossRef CAS.
- L. Kong, Z. Jiang, T. Xiao, L. Lu, M. O. Jones and P. P. Edwards, Chem. Commun., 2011, 47, 5512–5514 RSC.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS.
- X. Bo, G. Li, X. Qui, Y. Xue and L. Li, J. Solid State Chem., 2007, 180, 1038–1044 CrossRef CAS.
- J. S. Jang, P. H. Borse, J. S. Lee, O. S. Jung, C. R. Cho, E. D. Jeong, M. G. Ha, M. S. Won and H. G. Kim, Bull. Korean Chem. Soc., 2009, 30, 1738–1742 CrossRef CAS.
- B. D. Cullity, Elements of X-ray Diffraction, Addison-Wesley Formula Publishing, 2nd edn Search PubMed.
- A. Kudo and I. Mikami, Chem. Lett., 1998, 1027–1028 CrossRef CAS.
- R. Dom, R. Subasri, K. Radha and P. H. Borse, Solid State Commun., 2011, 151, 470–473 CrossRef CAS.
- N. Kislov, S. S. Srinivasan, Y. Emirov and E. K. Stefanakos, Mater. Sci. Eng., B, 2008, 153, 70–77 CrossRef CAS.
- J. Tauc, R. Grivorovici and A. Vancu, Phys. Status Solidi B, 1966, 15, 627–637 CrossRef CAS.
- A. Lagashetty, V. Hanvanoor, S. Basavaraja, S. D. Balaji and A. Venkataraman, Sci. Technol. Adv. Mater., 2007, 8, 484–487 CrossRef CAS.
- A. S. Vanetsev, A. E. Baranchikov and Y. D. Tret'Yakov, Dokl. Chem., 2008, 418, 34–36 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ra21910g |
| This journal is © The Royal Society of Chemistry 2012 |