Significance of dimer models describing physical properties in a triclinic solid of tin(II) phthalocyanine†
Michinori
Sumimoto
*a,
Teruyuki
Honda
b,
Yukio
Kawashima
c,
Kenji
Hori
a and
Hitoshi
Fujimoto
*b
aGraduate School of Science and Engineering, Yamaguchi University, Tokiwadai, Ube 755-8611, Japan. E-mail: sumimoto@yamaguchi-u.ac.jp
bDepartment of Chemistry, Faculty of Science, Kumamoto University, 2-39-1 Kurokami, Kumamoto 860-8555, Japan. E-mail: fuji@aster.sci.kumamoto-u.ac.jp
cDepartment of Chemistry, Graduate School of Sciences, Kyushu University, 6-10-1 Hakozaki, Higashiku, Fukuoka 812-8581, Japan
First published on 25th October 2012
Abstract
In order to study the solid-state electronic structures for ground and excited states of tin phthalocyanine (SnPc), we extracted two types of dimers and a trimer as model systems from the crystal structure of the triclinic polymorph. One of the dimers had a convex configuration between two SnPc molecules, and the other took a concave one. The trimer consisted of both configurations. The molecular geometries, electronic structures, and excitation energies of the concave- and convex-type dimers and the trimer were investigated using the M06 method of the density functional theory (DFT). The geometries of these model systems were optimized under C2h molecular symmetry for the dimers and under C1 for the trimer. The distances between two Sn atoms were optimized to be 6.805 and 6.374 Å for concave- and convex-type dimers, respectively. The corresponding distances in the trimer were calculated to be 6.959 and 6.457 Å, respectively. The Sn-Sn distances calculated for the dimers and trimer were consistent with the experimental values for the crystal. The energy diagrams of these model systems imply that each molecular orbital of an SnPc monomer forms the electron-energy band with a relatively large bandwidth due to strong intermolecular interactions in the solid state. The time-dependent DFT calculation for the trimer reproduced well the observed absorption spectrum in the solid state. It should be noted that a sum of the excited states calculated for the two types of the dimers reproduces the calculation results for the trimer well. From these results, it would be concluded that the electronic structure of SnPc for the solid state can be investigated by using these two dimers. The vertical values of the ionization potential (IP) were calculated with the optimized structures for the single molecule, two dimers and trimer of SnPc by the ΔSCF method. The first IPs decreased about 0.6 eV from monomer to trimer.
Introduction
Phthalocyanine compounds with the formula of MPc (Pc = phthalocyaninato anion C32H16N82–, M = H2 or divalent metals) have been subjects of a vast amount of study both theoretically and experimentally, because of their characteristic properties such as strong ultraviolet-visible absorption, highly symmetrical structure, small ionization potential, and their potential possibilities of industrial applications owing to their catalytic and semiconducting properties. MPcs have a macrocyclic ring with a large π-conjugated aromatic system consisting of four benzoisoindole units with a highly symmetrical square planar D4h structure in general.1–4Among many phthalocyanine compounds, tin(II) phthalocyanine (SnPc) has a unique molecular structure like a shuttlecock with a C4v symmetry.5,6 SnPc has gained considerable attention due to commercial applications such as organic semiconductors,7 organic materials for nonlinear optical devices,8,9 precursors for a new class of electrically conductive polymers,10 precursors for organic thin films of various properties,11 and as corrosion inhibitors.12 The protruding central atom makes SnPc derivatives interesting for a possible application as a single-molecular storage device.13
The triclinic polymorph has been reported in powder samples of SnPc.5,6Scheme 1 shows the mutual arrangement of molecules seen from the direction parallel to the Pc plane. One can see that the Sn atom is out of the convex molecular plane (convex-type) and the other is the concave molecular plane (concave-type).5 Interestingly, the absorption spectrum of the triclinic film is quite different from isolated SnPc monomer.6
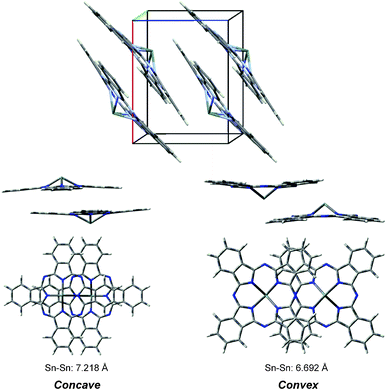 | ||
| Scheme 1 The triclinic polymorph of SnPc. | ||
Many efforts have been made on giving an explanation for the electronic structures of MPcs by theoretical calculations. The density functional theory (DFT) calculations using the local density approximation have been applied to investigate the geometric and the electronic structures of several MPc compounds.14–24 The time-dependent DFT (TD-DFT) has been employed efficiently to discuss the absorption spectra of SnPc, PbPc, and their derivatives.18,22 These works show that DFT methods are efficient, reliable and effective to investigate the molecular and electronic structures of MPcs for the present.
Recently, we attempted to give some explicit explanations for differences in the physical properties depending on polymorphs from the molecular geometries and electronic structures of a isolated LiPc molecule and its dimers, which correspond to the three polymorphs of the LiPc solids, by DFT methods.25–28 This approach successfully reproduced the experimental results of the molecular structures and the spin states for these three polymorphs of LiPc.26,27 We also explained qualitatively the differences in the absorption spectra of the three LiPc polymorphs from the ground and excited states electronic structures of the three dimers.28 This might show a possibility that the physical properties of the molecular solid can be investigated by using the electronic structures of the adequately chosen models with a small number of molecules such as dimers.
In this study, we will present the results from TD-DFT calculations for two model dimers (concave- and convex-type) and one model trimer of SnPc. The excited states were calculated using the optimized geometries of the single molecule, the two model dimers and the one model trimer of SnPc. The obtained results were inspected by comparison with the experimental results for the molecular structure in the crystalline solid and for the electronic absorption spectra in solution and in the solid state. One of the main purposes of this study is to inspect possibilities of a theoretical investigation into the physical properties of the molecular solids using model systems with a small number of molecules.
Experimental section
Theoretical calculations
Geometry optimization and the excitation energies of the monomer, two dimers (concave- and convex-types) and the trimer for SnPc were carried out using the DFT and TD-DFT29–31 methods, where the M06 functional32 was used for the exchange–correlation term. It has been shown that the M06 functional gave the most reliable results for the weakly bound system including a dispersion interaction.27,28 For constructing our model systems of crystal structures, the geometries for the two types of SnPc dimers were optimized under the following conditions: (1) Each SnPc moiety is optimized under constraint to keep C4v symmetry; (2) The distance and the tilt angle between the SnPc moieties were optimized simultaneously with SnPc dimers under constraint to keep C2h symmetry. For the trimer, we calculated the geometry with each SnPc constrained to keep C4v symmetry. The main purpose of this study is to give insight into the electronic structure of the triclinic solid; therefore, we only investigated the trimer with both configurations of the above two dimers.For the trimer geometry, all the calculated harmonic frequencies were confirmed to be real ones. On the other hand, both dimer models had one vibration mode with small negative frequency. Our main purpose is to reproduce molecular orientation in the triclinic solid in order to investigate its electronic structure; therefore, we proceeded with these geometries.
In these calculations, the following basis sets were employed. The LANL2DZ(d, p) basis set was used to represent the valence electrons of Sn, where the effective core potentials (ECPs) were employed to replace core electrons. For C, N and H atoms, the usual 6-311G(d) basis sets were employed. The Gaussian09 program package33 was used for these calculations. All molecular structures and contour maps of the molecular orbitals were drawn with the GaussView program package.34
Experiments
The commercially available powdery sample of SnPc was purified by vacuum sublimation just before use. The electronic absorption spectra of SnPc were measured for a chloroform solution and for a thin film. The thin film of SnPc was prepared by vapor deposition on a quartz substrate kept at room temperature under vacuum of 10−3 Pa. The deposition rate and the film thickness were monitored with a quartz thickness monitor, and were controlled at 1 nm min−1 and about 40 nm, respectively. Characterization of the evaporated film was performed by infrared and Raman spectra after the absorption measurements. The X-ray powder pattern was similar to the reported one,6 and this showed that the evaporated thin film of SnPc took a triclinic system (see Fig. S1 of the ESI†).Results and discussion
Optimized geometries and electronic structures of two SnPc dimers and trimer
In a previous paper,22 we reported a detailed study on the geometric and electronic structures of the SnPc monomer using the B3LYP calculation. Here, we would like to summarize the characteristic features of the electronic structures of the two SnPc dimers and the trimer for the sake of the discussion on their excited states. Fig. 1 illustrates the optimized structures of the concave- and convex-type SnPc dimers and the trimer using the M06 method. The relative Gibbs free energies of the two optimized dimers were estimated to be much lower than the sum of the independent monomers by about 8–10 kcal mol−1 at 298.15 K. The trimer also showed much lower value than the sum total of three monomers by 23.4 kcal mol−1 at 298.15 K. The total energies of dimers and trimer were less than the total sum of each consisting components. These data clearly showed that the stacking form would be preferable for SnPc units. As discussed in recent reports,23,27,28 the stabilization of the accumulation would come from the long-range interaction. The optimized geometries of the dimers had a C2h symmetry, and the distances between two Sn atoms were evaluated to be 6.805 and 6.374 Å for the concave- and convex-type dimers, respectively. The Sn–Sn distances in the dimers were similar to the observed values of 7.218 and 6.692 Å for the triclinic solid.5 Moreover, the corresponding distances in the trimer were calculated to be 6.959 and 6.457 Å, respectively. These values for the trimer became closer to the observed values than those for the dimers, and the differences between the observed and calculated values may be due to the effects of the crystal packing.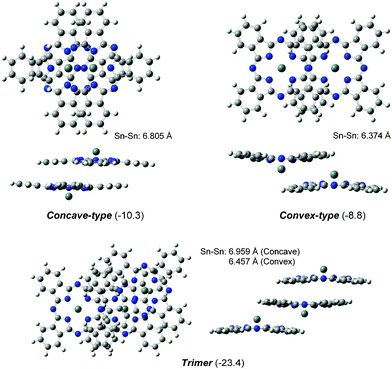 | ||
| Fig. 1 Optimized geometries of the concave- and convex-type dimers and the trimer of SnPc. Relative Gibbs free energies (kcal mol−1) to the sum of the monomers are in parentheses. | ||
Fig. 2 compares the energy diagrams of the monomer, two types of dimers and trimer in the region of the highest occupied (HOMO) and the lowest unoccupied (LUMO) molecular orbitals. The orbital symmetries are labelled under the C4v, C2h and C1 symmetry groups for the monomer, the dimers, and the trimer, respectively. All orbitals below a dotted line are occupied, and the corresponding orbitals among three model systems are combined with dashed lines. The result for the monomer was the same as those obtained by B3LYP calculation.22
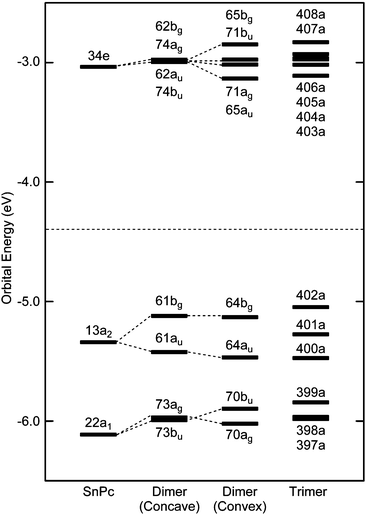 | ||
| Fig. 2 Orbital energies near the HOMO and LUMO for the monomer, the concave- and convex-type dimers, and the trimer of SnPc. The orbital symmetries are labeled under the C4v, C2h, C2h and C1 point groups for the monomer, the concave- and convex-type dimers, and the trimer of SnPc, respectively. For the SnPc monomer, the 134th MO is HOMO. The orbitals below the dotted line are occupied. | ||
The atomic contributions to the molecular orbitals are shown in Fig. S2, S3, and S4 of the ESI† for the concave- and convex-type dimers and the trimer, respectively. The HOMO of SnPc has a large contribution from carbon atoms of the inner C8N8 ring,22 and splits into the HOMO and HOMO−1 in dimers through anti-bonding and bonding interactions, respectively. These intermolecular orbital interactions of both dimers are limited in the region of two carbon atoms in the inner C8N8 ring; therefore, the splitting between the HOMO and HOMO−1 in the dimers would be small compared to the splitting due to the intra-molecular interaction in the double-decker type phthalocyanines.
Their general aspects are as follows. The orbitals of the concave- and convex-type dimer were generated from two orbitals of the SnPc moieties through in-phase or out-of-phase combinations. In the case of the trimer, the non-bonding type orbitals existed additionally in the middle of these two orbitals. The energy differences between HOMO and HOMO−1 were estimated to be about 0.30 and 0.34 eV in concave- and convex-type dimers, respectively. For trimer, these two molecular orbitals corresponded to HOMO and HOMO−2, respectively, and the difference was about 0.43 eV. These values are similar to the intermolecular bonding and anti-bonding orbital splitting of 0.35 eV in HOMO state for a bilayer film of lead phthalocyanine,35 which has the same shuttlecock molecular structure as SnPc. On the other hand, we have reported theoretically22 and experimentally36 that bis(phthalocyaninato)tin(IV) (SnPc2) shows a large splitting between HOMO and HOMO−1 of about 0.8 eV due to intra-molecular interactions. Comparing the results for SnPc and SnPc2, it would be concluded that limited orbital overlap in the SnPc dimers results in a smaller splitting than SnPc2. However, this splitting is still large compared to typical molecular solids. The energy diagrams of these model systems would imply that each molecular orbital of the SnPc monomer forms the electron-energy band with a relatively large bandwidth due to strong intermolecular interactions in the solid state. This might cause the exited states of SnPc to differ between isolated molecule and in the solid state.
Fig. 2 also shows the exchange repulsion in both of the dimers, where the stabilization of the HOMO−1 is smaller than the destabilization of HOMO as compared to HOMO of the monomer. As discussed in the recent papers,22,27,28 the London-type long-range dispersion interaction would play a crucial role in the weakly bound systems. In the present system, the long-range interaction such as a van der Waals interaction may also act as a driving force of accumulation.
Electronic absorption spectra of two SnPc dimers and the trimer
We calculated the excitation energies of two SnPc dimers and the trimer using the TD-DFT method. The optically allowed excited states obtained by the TD-M06/6-311G(d) method are shown as vertical lines for monomer, concave- and convex-type dimers, and the trimer in Fig. 3. Since the trimer was too large to complete the TD-DFT calculation in the full region under investigation of this study, we calculated the excited states only in the Q-band region below 2.4 eV. The height of each vertical line represents the oscillator strength of excitation. The solid line of each panel shows the theoretical absorption spectrum obtained by convoluting each excited state with a Gaussian function, where the FWHM was assumed to be 0.06 eV and the oscillator strength was used as the pre-exponential factor. The experimentally obtained absorption spectra for the thin film are also shown in the figure. The excitation energies of the dimers are summarized in Tables 1 and 2 for the optically-allowed excitations with oscillator strengths over 0.05. For the trimer, the values are compiled in Table S1 of the ESI†. The important components of the one electron transition, in which the coefficient is larger than 0.3, are also presented. The excitation energies and the important components for all optically allowed states of the dimers are shown in Tables S2 and S3 of the ESI.†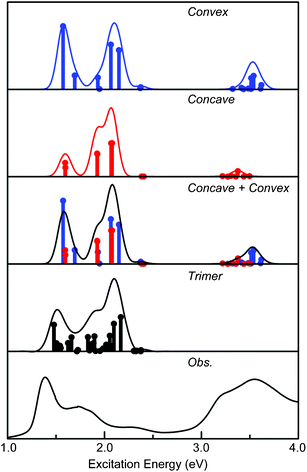 | ||
| Fig. 3 Excited states of the two dimers and the trimer of SnPc obtained by the TD-M06 calculation. The vertical line depicts the oscillator strength of each state. The theoretical absorption spectra were obtained by convoluting each excited state with a Gaussian function. The FWHM was assumed to be 0.06 eV and the oscillator strength was used as the pre-exponential factor. | ||
| State | Main configuration (|C| ≥ 0.30) | E a | f b | p c | |
|---|---|---|---|---|---|
| a Excitation energy in eV. b Oscillator strength. c Transition moment direction. | |||||
| C 2h | 11Bu | +0.65(61bg → 62au) [85%] | 1.59 | 0.106 | x+y |
| 11Au | +0.65(61bg → 74bu) [85%] | 1.60 | 0.069 | z | |
| 21Bu | +0.34(73bu → 74ag) [23%] −0.37(73ag → 74bu) [27%] +0.46(61au → 62bg) [42%] | 1.92 | 0.177 | x+y | |
| 21Au | −0.35(73bu → 62bg) [25%] −0.37(73ag → 62au) [27%] +0.46(61au → 74ag) [42%] | 1.93 | 0.172 | z | |
| 31Bu | −0.32(73bu → 74ag) [20%] −0.37(73ag → 74bu) [27%] +0.45(61au → 62bg) [41%] | 2.07 | 0.255 | x+y | |
| 31Au | −0.31(73bu → 62bg) [19%] −0.37(73ag → 62au) [27%] +0.45(61au → 74ag) [41%] | 2.08 | 0.256 | z | |
| 13Bu | −0.40(61au → 74ag) [32%] +0.61(61bg → 74bu) [74%] | 0.83 | 0.000 | — | |
| State | Main configuration (|C| ≥= 0.30) | E a | f b | p c | |
|---|---|---|---|---|---|
| a Excitation energy in eV. b Oscillator strength. c Transition moment direction. | |||||
| C 2h | 11Bu | +0.69(64bg → 65au) [95%] | 1.57 | 0.483 | x+y |
| 11Au | +0.67(64bg → 71bu) [90%] | 1.69 | 0.106 | z | |
| 21Au | +0.48(70ag → 65au) [46%] −0.35(70bu → 65bg) [25%] −0.37(64au → 71ag) [27%] | 1.93 | 0.091 | z | |
| 31Au | +0.34(70ag → 65au) [23%] +0.55(64au → 71ag) [61%] | 2.07 | 0.342 | z | |
| 31Bu | +0.66(64au → 65bg) [87%] | 2.15 | 0.300 | x+y | |
| 81Bu | +0.48(69bu → 71ag) [46%] | 3.51 | 0.086 | x+y | |
| 91Bu | −0.33(65ag → 71bu) [22%] +0.41(65bu → 71ag) [34%] | 3.54 | 0.103 | x+y | |
| 13Bu | −0.34(64au → 65bg) [23%] +0.64(64bg → 65au) [82%] | 0.78 | 0.000 | — | |
The thin solid film of SnPc exhibited two absorption features at 1.40 and 1.73 eV with a weak shoulder around 1.9 eV and a small hump around 2.3 eV in the typical Q-band region. In the B-band region, an intense broad absorption band with two structures around 3.22 and 3.55 eV was observed. These spectral features are completely different from those observed in solution.
The ground states of both concave- and convex-type dimers were calculated as state symmetry of 1Ag under C2h point group; therefore, the optically allowed excited states should belong to either 1Au or 1Bu state symmetry. By contrast, all excited states were symmetry allowed for the trimer. As shown by the simulated spectra, several excited states may group together and form an absorption band. Usually, excitation energies to Au states and the corresponding Bu states would have similar values and contribute pairs to the absorption spectrum. In the concave-type dimer, the first two excited states, 1Bu and 1Au, would form the first group around 1.6 eV, and they consisted mainly of transitions from HOMO to LUMO and to LUMO+1 for 1Au and 1Bu states, respectively. The second (2Bu and 2Au) and third (3Bu and 3Au) groups were calculated around 1.9 and 2.1 eV, respectively. These groups correlated with the molecular orbitals, which contained a large contribution from the central metal. These excited states would be assigned to have both the π–π* and the metal (5s of Sn) to ligand charge transfer (MLCT) characters. The excited states of the fourth group (4Au and 4Bu) were calculated around 2.4 eV and also corresponded to the mixed state of the π–π* and MLCT transitions, which would be observed as an absorption band with a small intensity. The Au and Bu states of each group had similar excitation energies. In the convex-type dimer, the excited states fell into several groups and contributed to the absorption spectrum as the concave-type dimer. The excitation energy differences between the concave- and convex-type dimers have arisen mainly from the orbital energies. Especially, the energies of the orbitals, which originated from the LUMO and HOMO−1 of the monomer, would affect strongly on the differences in the excitation energies of these dimers.
It has been reported5 that the unit cell of the triclinic SnPc crystal consists of two different configurations between two adjacent molecules, the concave and convex types. To simulate the absorption spectrum of the SnPc crystal, we performed TD-M06 calculation for the trimer, which contains configurations of both type of dimers. The TD-M06 calculation results of the trimer reproduced the observed absorption spectrum of the crystal well, as shown in Fig. 3. Moreover, it should be noted that the theoretical absorption spectrum for the trimer is well simulated by the sum of the excited states of concave- and convex-type dimers. These results clearly showed that the excited states and the excitation energy of the crystalline solid can be investigated sufficiently by using the model dimers chosen appropriately. In passing, information of the spin forbidden states is also expected to be important for experiments and applications of the material; therefore, the first triplet states of two dimers and the trimer were calculated and the results are compiled in Tables 1, 2 and S1, ESI.† The first triplet states of the systems calculated in the present study had a HOMO–LUMO transition as the main configuration, and will be observed in the near infrared region around 0.8 eV.
Ionization potentials of two SnPc dimers and trimer
The vertical values of the ionization potential (IP) were calculated with the optimized structures for the isolated molecule, two dimers and trimer of SnPc by the ΔSCF method. The obtained values are summarized in Table 3, compared with the observed value by the solid-state photoelectron spectroscopy. The IP value of the SnPc monomer was calculated to be 6.26 eV, which was very similar to the value obtained by the B3LYP method.22 The IP values of concave- and convex-type dimers were calculated to be 5.87 and 5.84 eV, respectively, and that of the trimer was 5.68 eV. This reduction in the first ionization potential would be strongly correlated to the intermolecular interaction, as discussed above for the energy diagrams of dimers and trimer. The HOMO of each SnPc molecule interacts and splits into two and three molecular orbitals in dimers and trimer, respectively. As the result, energies of HOMOs for dimers and trimer increase. This implies the band formation in the solid state, and the bandwidth originated from HOMO of the SnPc molecule can be estimated to be about at least 0.5 eV from the energy splitting between the HOMO and the HOMO−1 of the dimers. In the case of the usual molecular solid, the bandwidth is thought to be as small as about 0.1 eV. This indicates that there exists a strong intermolecular interaction in the solid of SnPc, which corresponds to the origin of the difference in absorption of SnPc in solution and in solid.| Calculationa | Experimentb | |
|---|---|---|
| a Vertical ionization potential by the ΔSCF. b The value was obtained for the SnPc thin film by the solid state photoelectron spectroscopy. | ||
| Compound | I P | I P |
| SnPc | 6.26 | 5.72 |
| Concave-type dimer | 5.87 | — |
| Convex-type dimer | 5.84 | — |
| Trimer | 5.68 | — |
Conclusions
We reported optimized geometries, electronic structures, and excitation energies for concave- and convex-type dimers and trimer of SnPc using M06 method. The geometries of two dimers and the trimer were optimized under C2h and C1 molecular symmetries, respectively.For concave- and convex-type dimers, the distances between two Sn atoms were estimated to be 6.805 and 6.374 Å, respectively. The corresponding distances in the trimer were calculated to be 6.959 and 6.457 Å, respectively. The Sn–Sn distances in dimers and trimer were very similar to the experimental values for those of the triclinic crystal structure.
The excitation energies of two dimers and trimer were calculated by TD-DFT method. TD-DFT calculation of the trimer reproduced the observed absorption spectrum of the triclinic crystal well. It should be mentioned with interest that the sum of the excited states of two dimers simulates the observed absorption spectrum for the solid state of SnPc well. From these results, we conclude that the excitation energies of the crystal can be investigated by using model dimers chosen appropriately.
The vertical values of IP were calculated with the optimized structures of the free molecule, two dimers and the trimer of SnPc by the ΔSCF method. The first ionization potentials of the trimer decreased by about 0.6 eV from the monomer, which indicates the strong interaction between the consisting SnPc monomers. From these values, we discussed the valence electron band of SnPc in the solid state.
Acknowledgements
This research was partially supported by TRANSITION STATE TECHNOLOGY CO. LTD., CEO: Toru Yamaguchi, Ph. D., the chemical venture in Japan.References
- A. B. P. Lever, Adv. Inorg. Chem. Radiochem., 1965, 7, 28 CrossRef.
- (a) W. R. Scheidt and W. Dow, J. Am. Chem. Soc., 1977, 99, 1101 CrossRef CAS; (b) J. F. Kirner, W. Dow and W. R. Scheidt, Inorg. Chem., 1976, 15, 1687 CrossRef.
- T. Kobayashi, F. Kurokawa, N. Uyeda and E. Suito, Spectrochim. Acta, 1970, 26A, 1305 CrossRef.
- (a) E. A. Lukyanets and V. N. Nemykin, J. Porphyrins Phthalocyanines, 2010, 14, 1 CrossRef CAS; (b) V. N. Nemykin and E. A. Lukyanets, ARKIVOC, 2010, i, 136 Search PubMed.
- R. Kubiak and J. Janczak, J. Alloys Compd., 1992, 189, 107 CrossRef CAS.
- A. Yamashita, S. Matsumoto, S. Sakata, T. Hayashi and H. Kanbara, J. Phys. Chem. B, 1998, 102, 5165 CrossRef CAS.
- S. R. Forrest, Chem. Rev., 1997, 97, 1793 CrossRef CAS.
- Y. Imanishi, S. Ishihara and T. Hamada, Mol. Cryst. Liq. Cryst. Sci. Technol. Sect. A, 1998, 315, 413 CrossRef CAS.
- Y. Imanishi and S. Ishihara, Thin Solid Films, 1998, 331, 309 CrossRef CAS.
- C. W. Dirk, T. Inabe, K. F. Schoch Jr. and T. J. Marks, J. Am. Chem. Soc., 1983, 105, 1539 CrossRef CAS.
- N. Papageorgiou, E. Salomon, T. Angot, J.-M. Layet, L. Giovanelli and G. Le Lay, Prog. Surf. Sci., 2005, 77, 139 CrossRef.
- H. I. Beltran, R. Esquivel, M. Lozada-Cassou, M. A. Dominguez-Aguilar, A. Sosa-Sanchez, J. L. Sosa-Sanchez, H. Hoepfl, V. Barba, R. Luna-Garcia, N. Farfan and L. S. Zamudio-Rivera, Chem.–Eur. J., 2005, 11, 2705 CrossRef CAS.
- K. Walzer and M. Hietschold, Surf. Sci., 2001, 471, 1 CrossRef CAS.
- P. N. Day, Z. Wang and R. Pacher, J. Mol. Struct. (THEOCHEM), 1998, 33, 455 Search PubMed.
- C. Ruan, V. Mastryukov and M. Fink, J. Chem. Phys., 1999, 111, 3035 CrossRef CAS.
- J. D. Baran and J. A. Larsson, Phys. Chem. Chem. Phys., 2010, 12, 6179 RSC.
- (a) Y. Zhang, X. Zhang, Z. Liu, Y. Bian and J. Jiang, J. Phys. Chem. A, 2005, 109, 6363 CrossRef CAS; (b) Y. Zhang, X. Zhang, Z. Liu, H. Xu and J. Jiang, Vib. Spectrosc., 2006, 40, 289 CrossRef CAS.
- Y. Zhang, X. Cai, X. Zhang, H. Xu, Z. Liu and J. Jiang, Int. J. Quantum Chem., 2007, 107, 952 CrossRef CAS.
- (a) N. Papageorgiou, Y. Ferro, E. Salomon, A. Allouche, J. M. Layet, L. Giovanelli and G. Le Lay, Phys. Rev. B: Condens. Matter, 2003, 68, 235105 CrossRef; (b) E. Salomon, N. Papageorgiou, Y. Ferro and J. M. Layet, Thin Solid Films, 2004, 466, 259 CrossRef CAS.
- T. Strenalyuk, S. Samdal and H. V. Volden, J. Phys. Chem. A, 2008, 112, 10046 CrossRef CAS.
- (a) M. Sumimoto, Y. Kawashima, K. Hori and H. Fujimoto, Spectrochim. Acta, Part A, 2008, 71, 286 CrossRef; (b) M. Sumimoto, Y. Kawashima, K. Hori and H. Fujimoto, Dalton Trans., 2009, 5737 RSC; (c) R. Murdey, M. Bouvet, M. Sumimoto, S. Sakaki and N. Sato, Synth. Met., 2009, 159, 1677 CrossRef CAS.
- M. Sumimoto, T. Honda, Y. Kawashima, K. Hori and H. Fujimoto, Dalton Trans., 2012, 41, 7141 RSC.
- (a) N. Marom and L. Kronik, Appl. Phys. A: Mater. Sci. Process., 2009, 95, 159 CrossRef CAS; (b) N. Marom and L. Kronik, Appl. Phys. A: Mater. Sci. Process., 2009, 95, 165 CrossRef CAS; (c) T. Körzdörfer, S. Kümmel, N. Marom and L. Kronik, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 201205 CrossRef; (d) N. Marom, A. Tkatchenko, M. Scheffler and L. Kronik, J. Chem. Theory Comput., 2010, 6, 81 CrossRef CAS.
- V. N. Nemykin, R. G. Hadt, R. V. Belosludov, H. Mizuseki and Y. Kawazoe, J. Phys. Chem. A, 2007, 111, 12901 CrossRef CAS.
- T. Kimura, M. Sumimoto, S. Sakaki, H. Fujimoto, Y. Hashimoto and S. Matsuzaki, Chem. Phys., 2000, 253, 125 CrossRef CAS.
- M. Sumimoto, S. Sakaki, S. Matsuzaki and H. Fujimoto, Dalton Trans., 2003, 31 RSC.
- (a) M. Sumimoto, Y. Kawashima, D. Yokogawa, K. Hori and H. Fujimoto, J. Comput. Chem., 2011, 32, 3062 CrossRef CAS; (b) M. Sumimoto, Y. Kawashima, D. Yokogawa, K. Hori and H. Fujimoto, Int. J. Quantum Chem., 2012 DOI:10.1002/qua.24072.
- M. Sumimoto, D. Yokogawa, Y. Kawashima, K. Hori and H. Fujimoto, Spectrochim. Acta, Part A, 2012, 91, 118 CrossRef CAS.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454 CrossRef CAS.
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439 CrossRef CAS.
- R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- R. Dennington, T. Keith and J. Millam, Gauss View, Version 5, Semichem Inc., Shawnee Mission KS, 2009 Search PubMed.
- S. Kera, H. Fukagawa, T. Kataoka, S. Hosoumi, H. Yamane and N. Ueno, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 121305(R) CrossRef.
- T. Honda, Y. Ohya, M. Sumimoto, T. Moriguchi, K. Hori and H. Fujimoto, Chem. Phys., 2012, 407, 110 CrossRef.
Footnote |
| † Electronic Supplementary Information (ESI) available: Cartesian coordinates for all species and molecular orbitals and all calculated excitation energies for all species and the X-ray powder pattern of the evaporated thin film of SnPc in a triclinic system. See DOI: 10.1039/c2ra21509h |
| This journal is © The Royal Society of Chemistry 2012 |