DOI:
10.1039/C2RA22549B
(Communication)
RSC Adv., 2012,
2, 12718-12723
A nanocomposite gel based on 1D coordination polymers and nanoclusters reversibly gelate water upon heating†
Received
17th October 2012
, Accepted 23rd October 2012
First published on 23rd October 2012
Abstract
A stable nanocomposite metallogel was obtained by the addition of [Na(L)(H2O)]·2H2O (HL = 4,6-bis(2-pyridyl)-1,3,5-triazin-2-ol) to an aqueous solution of Cu2(OAc)4·2H2O in a molar ratio of Cu/L = 4 (5.6 wt% gelator concentration), from which 1D coordination polymers [Cu2L(μ-OAc)2(μ-OH)] (1) and coordination nanoclusters {[Cu9L4(OAc)7(OH)5]2+}2 (3) were afforded. The nanoclusters 3 stabilize the 1D coordination polymers 1 and inhibit crystallization of 1 from the system, generating a coordination-based heat-set nanocomposite hydrogel at room and higher temperatures that flows upon cooling to 4 °C, in sharp contrast to most supramolecular gels. Reversible thermo and pH responsive solution–gel phase transitions were observed. The results described herein may open a novel avenue for functional coordination nanocomposite materials.
1 Introduction
Hydrogels,1 environmentally-friendly soft materials, have attracted intense attention due to their potential applications in tissue engineering,2 vehicles for controlled drug release,2b,3 pollutant detection and removal.4 Metallogels, assembled from organic ligands and metal ions, provide opportunities to introduce the properties of metals, such as magnetic, catalytic, spectroscopic and redox behavior into soft materials.5 They are promising responsive materials towards various stimuli including sonification,6 heat,7 pH,8 chirality,6b,f,9 mechanics6e,10 and reduction/oxidation.7e,11 Nanocomposite hydrogels, a combination of several components at the molecular level, is one of the most effective approaches for producing new materials with advanced properties,12 and those consisting of metal particles/clusters or clay embedded in organic polymers have been well developed.13 However, coordination-based composite hydrogels are still rare, though coordination polymers are also able to act as a host matrix for nanoscale objects of diverse compositions.14
Previous work has shown that the ligand 4,6-bis(2-pyridyl)-1,3,5-triazin-2-ol (HL) (see Scheme 1) is a multidentate ligand and has various coordination modes. Using this ligand, a cubic, nanosize cage cation [Co(H2O)6 ⊂ Co8(L)12]6+ has been successfully assembled in the presence of a Co(II) ion.15 When the hard metal ions were introduced into the reactions, 1D coordination polymers have been obtained.16 As part of an ongoing study, we extend the observation of the reaction of the ligand towards copper ions in various conditions. Surprisingly, when [Na(L)(H2O)]·2H2O or [Co(H2O)6 ⊂ Co8(L)12](NO3)6·12H2O was mixed with an aqueous solution of copper acetate, a green transparent gel was generated, from which crystals were obtained depending on the crystallization temperature.17 In this contribution, we report for the first time that a nanocomposite gel based on the HL ligand, containing a 1D coordination polymer and coordination nanocluster, has been discovered experimentally to gelate water selectively. Remarkably, the coordination nanoclusters stabilize the 1D coordination polymers to form a hydrogel that flows upon cooling, in sharp contrast to most supramolecular gels. The purpose of the research herein is to develop a better understanding of the mechanism of gel formation in nanocomposite materials and subsequently to elucidate the nature of their responsive behaviors.
 |
| | Scheme 1 The structures of 4,6-bis(2-pyridyl)-1,3,5-triazin-2-ol (HL). | |
2 Experimental
2.1 Materials and physical technique
The reagents and solvents employed were commercially available and were used as received without further purification. [Na(L)(H2O)]·2H2O and [Co(H2O)6 ⊂ Co8L12](NO3)6·12H2O was prepared according to previously reported procedures.15a,16 The C, H and N microanalyses were carried out with a Vario EL elemental analyzer. The FT-IR spectra were recorded from KBr pellets in the range of 400–4000 cm−1 on a Bruker TENSOR 27 spectrometer. Thermogravimetric data were collected on a Netzsch TGS-2 analyzer in a nitrogen atmosphere at a heating rate of 10 °C min−1. Powder X-ray diffraction patterns were recorded on a D/Max-2200 diffractometer with Cu–Kα radiation (λ = 1.5409 Å) at a scanning rate of 4° min−1 with 2θ ranging from 3° to 30°. Raman spectroscopy was measured in the backscattering geometry utilizing a Leica microscopy system equipped with a 50× objective in a Renishaw inVia spectrometer. The wavelength of the irradiating argon ion laser was 514.5 nm and the spectra resolution was 1 cm−1. The laser light was focused to a spot size of ca. 1 μm diameter on the sample surface and the power was less than 2 mW. SEM images were taken with a FEI Quanta 400 Thermal FE environmental scanning electron microscope. Samples were gold-coated prior to the SEM analysis. X-ray photoelectron spectroscopy (XPS) measurements were made on an ESCALAB250 machine with a monochromatic Al–Kα source and all the binding energies were referenced to the C 1s peak at 284.6 eV.
2.2 Typical procedure for the preparation of the metallogel
[Na(L)(H2O)]·2H2O (32.7 mg, 0.1 mmol) was added to a 2 mL aqueous solution of Cu2(OAc)4·2H2O (80.0 mg, 0.2 mmol) and the mixture was stirred for 5 min at room temperature. The gel samples used for the characterization were aged for an appropriate period at room temperature.
2.3 Synthesis of [Cu9L4(OAc)7(OH)5]2(NO3)4·32H2O (3·NO3)
[Co(H2O)6 ⊂ Co8L12](NO3)6·12H2O (200 mg, 0.047 mmol) and Cu2(OAc)4·2H2O (200 mg, 0.5 mmol) were dissolved into a mixture of distilled water (1 mL) and methanol (20 mL). The solution was allowed to concentrate slowly at 30 °C. After several hours, it gelated. The phase separation was observed after 4 days with plate-like green crystals. The crystals were carefully separated from the solution and their purity were confirmed by PXRD. The formula was confirmed by XPS, EA, TGA (see Figs. S1 and S2, ESI†). Yield: 137 mg, 49.6%. Anal. Calcd for C132H180Cu18N44O90: C 31.92, H 3.65, N 12.41; Found: C 32.11, H 3.608, N 12.46. FT-IR (KBr, cm−1): 3431(br), 1674(m), 1652(m), 1618(m), 1579(s), 1544(vs), 1514(vs), 1471(s), 1434(s), 1404(s), 1384(vs), 1028(m), 794(m).
2.4 Gel melting temperature (Tgel) determination
Thermotropic behaviors of the hydrogels were carried out by using the dropping ball method.18 A glass ball weighing 18 mg (diameter of ∼1.5 mm) was placed on the top of a 2 mL volume of the hot gel in a glass vial with a diameter of 1.7 cm. The vial was sealed. The gel was then slowly cooled and the temperature at which the ball reached the bottom of the vial was taken as the sol–gel transition temperature (Tgel).
2.5 X-ray crystallography
Diffraction intensities were collected at 123 K for 3·NO3 on a Bruker Smart Apex CCD diffractometer with graphite-monochromated Mo–Kα radiation (λ = 0.71073 Å). Absorption corrections were applied using SADABS.19 The structures were solved by direct methods and refined with the full-matrix least-squares technique using the SHELXS-97 and SHELXL-97 programs, respectively.20 Anisotropic thermal parameters were applied to all non-hydrogen atoms, except for the disordered nitrate ions and water molecules in 3·NO3. The organic hydrogen atoms were generated geometrically (C–H 0.96 Å). Efforts to locate and refine the solvent and nitrate peaks were in vain. The SQUEEZE routine was used to remove the scattering from the highly disordered solvent molecules and nitrate ions.21 The structure of 3·NO3 was then refined again using the new intensity data generated by SQUEEZE. The molecular formula of 3·NO3 was confirmed on the basis of charge balance, EA, TGA and XPS. Crystal data as well as the details of the data collection and refinement for the complex is summarized in Table 1.
| Formula |
C132H116N41O50Cu18 |
|
R
1 = ∑||Fo| − |Fc||/∑|Fo|, wR2 = [∑w(Fo2 − Fc2)2/∑w(Fo2)2]1/2. |
| Formula weight |
4220.38 |
| Crystal system |
Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
|
a/Å |
14.7198(7) |
|
b/Å |
19.2788(9) |
|
c/Å |
20.2209(10) |
|
α (°) |
113.400(1) |
|
β (°) |
106.960(1) |
|
γ (°) |
100.281(1) |
|
V/Å3 |
4745.7(4) |
|
Z
|
1 |
|
D
c/g cm−3 |
1.477 |
|
μ/mm−1 |
2.049 |
| Data/parameters |
18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 423/1088 423/1088 |
|
R
1 (I > 2σ) |
0.0450 |
|
wR
2 (all data) |
0.1307 |
| GOF |
1.000 |
| Δρmax/Δρmin (eÅ3) |
1.06/−0.90 |
3 Results and discussion
3.1 Gelation process and characterization
The metallogel was obtained by the addition of [Na(L)(H2O)]·2H2O to an aqueous solution of Cu2(OAc)4·2H2O in various molar ratios. The gelation only selectively occurred in water, not other organic solvents (DMSO, DMF, MeOH, EtOH, THF, MeCN, CH3COCH3, CHCl3 and HOCH2CH2OH). Moreover, only copper acetate has been found to induce gelation compared with other copper salts (Cu(NO3)2, CuCl2, CuSO4, Cu(ClO4)2). When the molar ratio of Cu to L is 4 (5.6 wt% gelator concentration), the metallogel formed and was stable in a sealed vial for at least a year as no phase separation was observed during this period at room temperature. This was sharply different from the previous observation at a molar ratio of Cu/L = 2.6 (6.5 wt%), where a green metastable gel was obtained at room temperature and two isomers of a 1D chain coordination polymer [Cu2L(μ-OAc)2(μ-OH)]n (1) and a tetranuclear macrocycle [Cu4L2(μ-OAc)4(μ-OH)2]·H2O (2·H2O) crystallized upon phase separation by cooling and heating, respectively.17 However, both 1 and 2·H2O have poor solubility in water and are not hydrogelators on their own or together due to the well-regulated organization in the crystalline material.
The aforementioned findings encouraged us to systematically observe the gel behaviors. Firstly, the thermal response of the gel was examined. Surprisingly, the gel turns into a fluid solution upon cooling below 4 °C (see Fig. 1), as opposed to conventional gels that are dissolved upon heating, indicating that it is a rare heat-set metallogel.7a,c The gel-to-solution transitions are reversible via a change of temperature. It is worth noting that the solution is stable at low temperature and different from that observed in the metastable gel,17 suggesting that the gel stability is very dependent on the Cu/L ratio and the gelator concentration (vide infra). Secondly, the gel is reversibly pH responsive. When HOAc was added to the gel at room temperature, it turned into a fluid solution. The reverse process was observed upon the addition of 25% NH3·H2O. Interestingly, the acidic solution gelated again upon heating at 60 °C and fluidified reversibly upon cooling to room temperature. This fact indicates that the gel stability also depends on the pH.
 |
| | Fig. 1 Stimuli-responsive behaviors and phase transitions of the metallogel. | |
To evaluate the gel properties, the relationship of the gel-to-solution transition temperature (Tgel) vs. the gelator concentration, amount of HOAc, ageing time and molar ratio of Cu/L were determined by the “dropping ball method”.18 As shown in Fig. 2, increasing the amount of HOAc raised the Tgel values in the range of experiments and the addition of more HOAc destroyed the gel. This observation suggests that the thermal stability of the gel can be tuned by pH. The Tgel values decreased with an increase of gelator concentration.‡ This behavior indicates that the stability of the gel networks increased with the gelator concentration.7e Similar cases were also observed for the Tgel value towards ageing time and the gel-to-solution transition time significantly depended on the ageing time. For example, the gel aged for one-year took about a week to liquefy whereas the gel aged for one-day only needed 10 min to form a solution at low temperature, demonstrating that ageing appears to significantly enhance the thermal stability of the gel.22 Interestingly, the gel was strengthened as the Cu/L molar ratio went up from 4 to 7, however, further increases of the Cu/L molar ratio destabilized the gel, indicating that the appropriate Cu/L ratio plays a crucial role in the change of the thermal stability of the gel (vide infra).
 |
| | Fig. 2 Phase diagram plotted in terms of Tgelvs. various factors. (a) The addition of HOAc (Cu/L = 4, 5.6 wt%, one-day-aged); (b) the change of gelator (Cu/L = 4, one-day-aged); (c) the aged time (Cu/L = 4, 4.2 wt%); (d) the change of the molar ratio of Cu/L (4.2 wt%, one-day-aged). | |
3.2 Chemical structure of the nanocomposite hydrogel
To gain a better understanding of the gel formation mechanism, we attempted to grow single crystals from the system at different temperatures. However, these efforts failed and only powder samples were afforded. We tried to get some clues from a related system and another metallogel was obtained when the cluster [Co(H2O)6 ⊂ Co8L12](NO3)6·12H2O was used as a starting material instead of [Na(L)(H2O)]·2H2O, and was added to a solution of Cu2(OAc)4·2H2O at room temperature. Interestingly, plate-like green crystals of [Cu9L4(OAc)7(OH)5]2(NO3)4·32H2O (3·NO3) were obtained from the metallogel after 4 days at room temperature. An X-ray diffraction study revealed that 3 is an eighteen-nuclear nanocluster with dimensions of 18.9 × 15.8 × 14.2 Å, connecting two tetranuclear cycles and two pentanuclear cycles via ten weak Cu–O bonds with distances from 2.21 to 2.40 Å (see Fig. 3 and the ESI†). Significantly, the dicopper [Cu2L2(μ-OH)(μ-OAc)2] core in 3 is the same as that previously observed in 1 and 2.17 In contrast to 1 and 2, 3·NO3 is soluble in water and methanol. Complex 3 was used to clarify the gel components (vide infra).
 |
| | Fig. 3 X-ray crystal structure of 3·NO3. The hydrogen atoms, solvent molecules and uncoordinated anions were omitted for clarity (Cu: lime; N: blue; O: red; C: grey). | |
On the other hand, to further probe the components in the solution and gel, the samples were freeze-dried to minimize the change in the components.§ A soft aerogel G was obtained by drying the gel (5.6 wt%, Cu/L = 4) that was aged for a year at room temperature. A loose block material S was prepared by freeze-drying the solution that was obtained from the aged gel by cooling for a week. Raman spectra of the dried samples were measured and compared with complexes Cu2(OAc)4·2H2O, 1, 2, and 3·NO3 (see Fig. S3 in the ESI†). The Raman spectra of G, S, 1, 2, and 3·NO3 are very similar in the range of 100–1660 cm−1, excepting some characteristic peaks of Cu2(OAc)4·2H2O which appear in G and S. The characteristic Raman peak at 1663 cm−1 for 1 and 1697 cm−1 for 3·NO3 were located in both S and G, however, the peak at 1673 cm−1 for 2 could not be observed in either of them, indicating that 1 and 3 may co-existed in both the solution and gel. Moreover, immersing the solid materials G and S in water both resulted in the reformation of hydrogel.
Furthermore, the materials S and G were washed with MeOH to remove the soluble species and the resultant powdery solids were denoted as S-MeOH and G-MeOH, respectively. Their IR spectra and powder XRD patterns are identical to that of complex 1 (see Fig. 4 and 5), indicating S-MeOH, G-MeOH and complex 1 have the same components. Moreover, the filtrate was concentrated and then investigated by IR. A similar IR spectrum to that of complex 3 was observed, excepting that two strong peaks at 1621 and 1432 cm−1 for Cu2(OAc)4·2H2O appear in the filtrate (see Fig. S4, ESI†). These also strongly support that complexes 1 and 3 are the main components in both the gel and the solution.
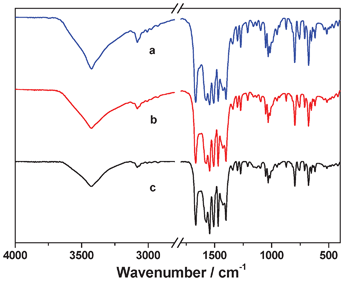 |
| | Fig. 4 Comparison of the IR spectra of complex 1 (a), S-MeOH (b) and G-MeOH (c). | |
 |
| | Fig. 5 Comparison of PXRD patterns of complex 1 (a), S-MeOH (b), and G-MeOH (c). | |
Complexes 1 and 3 were obtained in the molar ratio of Cu/L = 4, whereas complexes 1 and 2 were afforded in a molar ration of Cu/L = 2.6.17 The above experiments raise questions concerning the final products and the molar ratio of Cu/L. To answer this question, complex 3·NO3 was added to a solution of NaOAc. A gel was obtained after 5 min and then complex 2 crystallized out from the gel at room temperature after 3 h, indicating that 3 may be an intermediate species that can be converted into complex 2 in the presence of acetate. Comparing the X-ray crystal structures of 2 and 3, one can find that the molar ratio of Cu:L:OAc is 9:4:7 in 3 and 4:2:4 in 2. That is to say, the ratio of Cu/L in 3 is higher than that in 2, although the ratio of OAc/L in 3 is lower than that in 2. These results can explain why complex 3 was obtained under a large excess of Cu2(OAc)4·2H2O and 3·NO3 can be transformed into 2 in the presence of acetate. These observations also illustrate that an excess of Cu2(OAc)4·2H2O plays an important role in the formation of a stable gel. When the amount of Cu2(OAc)4·2H2O decreases, complex 3 may convert into complex 2, destabilizing the gel and leading to crystallization.17
3.3 Morphology of the nanocomposite hydrogel
To understand the mechanism for the unusual behavior of the heat-set metallogel, scanning electron microscopy (SEM) was used to observe the morphologies of solids S, S-MeOH, G, and G-MeOH at the microscopic level, providing an alternative view in the impact of molecular self-assembly. The SEM image of S reveals the presence of a plate-like structure with rods emerging on the surface (see Fig. 6a). However, a spongy network structure appears in aerogel G (see Fig. 6b). This is different from the observation in solid S. Most importantly, the SEM image of G-MeOH, obtained by washing aerogel G with MeOH, presents an entangled fibrillar network structure (see Fig. 6d), indicating that the 1D coordination polymers (1) (vide supra) entangle to form a host matrix that can be effective in trapping water molecules in the gel phase. Meanwhile, S-MeOH displays a linear rod-like structure (length, ca. 3 μm and width, ca. 1 μm). These results demonstrate that the fibrillar coordination polymers (1) weave into the host gel matrix and nanoclusters (3) may be embedded into the matrix, resulting in the spongy-like gel network.
 |
| | Fig. 6 SEM image of (a) S, (b) G, (c) S-MeOH, (d) G-MeOH. | |
3.4 Formation mechanism of the nanocomposite hydrogel
Based on the above results, a gelation mechanism has been proposed in Scheme 2. When the polydentate ligand [Na(L)(H2O)]·2H2O was added to an aqueous solution of Cu2(OAc)4·2H2O, 1D coordination polymers (1) and nanoclusters (3) were afforded. Together, they gelate water at room and high temperatures, in which the 1D coordination polymers might intertwist into a fibrillar network structure that acts as a host matrix which adopts the nanoclusters. This entangled interaction becomes stronger as the temperature increases and water molecules are entrapped among the polymeric network, leading to the formation of a robust hydrogel. However, the 1D coordination polymers are well dispersed at low temperatures, resulting in a flowing solution with low viscosity. It should be pointed out that nanocluster 3 plays an indispensable role in the formation of the hydrogel. The existence of nanocluster ions around the 1D coordination polymers may kinetically inhibit the crystallization of 1 from the gel or solution, considering the building blocks of the gel networks have no flexible branched group and their close packing consequentially leads to crystallization.
 |
| | Scheme 2 Schematic illustration of the formation of the heat-induced nanocomposite metallogel. | |
In fact, the heat-driven polymer aggregation model has been proposed for the reverse thermal gelation of polymers in water,23 where the polymers consist of a hydrophilic ethylene oxide dendritic exterior. Our current report extends the reverse thermal gelation concept to nanocomposite material on the basis of coordination polymers and nanoclusters, which has never been reported before. Unlike most of the thermo-induced gels that have long hydrophilic/hydrophobic chains,7,23 in our case, no flexible branched group appends on the 1D coordination polymer chains or around the nanoclusters, they gelate water molecules together upon heating and liquefy upon cooling. Here, the mechanism of gel formation is also different from that of heat-set coordination-based gels on the basis of the structural change in coordination polymers, in which tetrahedral triazole Co(II) species in the gel are transformed into octahedral species in solution upon cooling,7a and 1D Cu(I) coordination polymers in solution are transformed to a 2D network in the gel at high tempertures.7c
4 Conclusions
In summary, a coordination-based nanocomposite heat-set hydrogelator has been demonstrated for the first time, in which the nanoclusters (3) stabilize the 1D coordination polymers (1) and inhibit the crystallization of 1. The hydrogel is responsive to a number of stimuli including heat, pH and concentration. A nanocomposite gelation mechanism has been proposed to explain the rare characteristics of low viscosity at low temperatures and gel formation at high temperatures. The results described herein may open a novel avenue for functional nanocomposite coordination materials.
Acknowledgements
This work was supported by the NSFC (21071154, 20903121 and 21001031). We thank Mr. Zi-Long Wang for the SEM experiments.
References
-
(a) L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1217 CrossRef CAS;
(b) M. de Loos, B. L. Feringa and J. H. van Esch, Eur. J. Org. Chem., 2005, 3615–3631 CrossRef CAS;
(c) J. Kopecek, Biomaterials, 2007, 28, 5185–5192 CrossRef CAS;
(d) G. John, S. R. Jadhav, V. M. Menon and V. T. John, Angew. Chem., Int. Ed., 2012, 51, 1760–1762 CrossRef CAS.
-
(a) K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1879 CrossRef CAS;
(b) N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS;
(c) J. L. Drury and D. J. Mooney, Biomaterials, 2003, 24, 4337–4351 CrossRef CAS;
(d) Z. Yang, G. Liang, L. Wang and B. Xu, J. Am. Chem. Soc., 2006, 128, 3038–3043 CrossRef CAS.
-
(a) A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002–8018 CrossRef CAS;
(b) F. Zhao, M. L. Ma and B. Xu, Chem. Soc. Rev., 2009, 38, 883–891 RSC;
(c) N. A. Peppasa, P. Buresa, W. Leobandunga and H. Ichikawa, Eur. J. Pharm. Biopharm., 2000, 50, 27–46 CrossRef;
(d) J.-J. Zhang, W. Lu, R. W.-Y. Sun and C.-M. Che, Angew. Chem., Int. Ed., 2012, 51, 4882–4886 CrossRef CAS.
-
(a) N. Dave, M. Y. Chan, J. P.-J. Huang, B. D. Smith and J. Liu, J. Am. Chem. Soc., 2010, 132, 12668–12673 CrossRef CAS;
(b) O. Ozaya, S. Ekicia, Y. Barana, N. Aktasb and N. Sahiner, Water Res., 2009, 43, 4403–4411 CrossRef.
-
(a) F. Fages, Angew. Chem., Int. Ed., 2006, 45, 1680–1682 CrossRef CAS;
(b) M.-O. M. Piepenbrock, G. O. Lloyd, N. Clarke and J. W. Steed, Chem. Rev., 2010, 110, 1960–2004 CrossRef CAS;
(c) E. Carretti, M. Bonini, L. Dei, B. H. Berrie, L. V. Angelova, P. Baglioni and R. G. Weiss, Acc. Chem. Res., 2010, 43, 751–760 CrossRef CAS;
(d) D. D. Díaz, D. Kühbeck and R. J. Koopmans, Chem. Soc. Rev., 2011, 40, 427–448 RSC.
-
(a) T. Naota and H. Koori, J. Am. Chem. Soc., 2005, 127, 9324–9325 CrossRef CAS;
(b) N. Komiya, T. Muraoka, M. Iida, M. Miyanaga, K. Takahashi and T. Naota, J. Am. Chem. Soc., 2011, 133, 16054–16061 CrossRef CAS;
(c) S. Zhang, S. Yang, J. Lan, Y. Tang, Y. Xue and J. You, J. Am. Chem. Soc., 2009, 131, 1689–1691 CrossRef CAS;
(d) J. M. J. Paulusse, D. J. M. van Beek and R. P. Sijbesma, J. Am. Chem. Soc., 2007, 129, 2392–2397 CrossRef CAS;
(e) W. Weng, J. B. Beck, A. M. Jamieson and S. Rowan, J. Am. Chem. Soc., 2006, 128, 11663–11672 CrossRef CAS;
(f) X. Chen, Z. Huang, S.-Y. Chen, K. Li, X.-Q. Yu and L. Pu, J. Am. Chem. Soc., 2010, 132, 7297–7299 CrossRef CAS.
-
(a) K. Kuroiwa, T. Shibata, A. Takada, N. Nemoto and N. Kimizuka, J. Am. Chem. Soc., 2004, 126, 2016–2021 CrossRef CAS;
(b) H.-J. Kim, J.-K. Kim and M. Lee, Chem. Commun., 2010, 46, 1458–1460 RSC;
(c) X. de Hatten, N. Bell, N. Yufa, G. Christmann and J. R. Nitschke, J. Am. Chem. Soc., 2011, 133, 3158–3164 CrossRef CAS;
(d) Y. He, Z. Bian, C. Kang, Y. Cheng and L. Gao, Chem. Commun., 2010, 46, 3532–3534 RSC;
(e) D. Braga, S. d'Agostino, E. D'Amen and F. Grepioni, Chem. Commun., 2011, 47, 5154–5156 RSC;
(f) W. L. Leong, A. Y.-Y. Tam, S. K. Batabyal, L. W. Koh, S. Kasapis, V. W.-W. Yam and J. J. Vittal, Chem. Commun., 2008, 3628–3630 RSC;
(g) J. H. Lee, H. Lee, S. Seo, J. Jaworski, M. L. Seo, S. Kang, J. Y. Lee and J. H. Jung, New J. Chem., 2011, 35, 1054–1059 RSC;
(h) A. Blanazs, R. Verber, O. O. Mykhaylyk, A. J. Ryan, J. Z. Heath, C. W. Ian Douglas and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 9741–9748 CrossRef CAS.
-
(a) I. Odriozola, I. Loinaz, J. A. Pomposo and H. J. Grande, J. Mater. Chem., 2007, 17, 4843–4845 RSC;
(b) P. Bairi, B. Roy and A. K. Nandi, J. Mater. Chem., 2011, 21, 11747–11749 RSC;
(c) H. Lee, J. H. Lee, S. Kang, J. Y. Lee, G. John and J. H. Jung, Chem. Commun., 2011, 47, 2937–2939 RSC;
(d) Q. Wei and S. L. James, Chem. Commun., 2005, 1555–1556 RSC.
-
(a) T. Tu, W. Fang, X. Bao, X. Li and K. H. Dötz, Angew. Chem., Int. Ed., 2011, 50, 6601–6605 CrossRef CAS;
(b) J.-S. Shen, G.-J. Mao, Y.-H. Zhou, Y.-B. Jiang and H.-W. Zhang, Dalton Trans., 2010, 39, 7054–7058 RSC.
-
(a) T. D. Hamilton, D.-K. Bučar, J. Baltrusaitis, D. R. Flanagan, Y. Li, S. Ghorai, A. V. Tivanski and L. R. MacGillivray, J. Am. Chem. Soc., 2011, 133, 3365–3371 CrossRef CAS;
(b) W. Weng, Z. Li, A. M. Jamieson and S. J. Rowan, Macromolecules, 2009, 42, 236–246 CrossRef CAS;
(c) M.-O. M. Piepenbrock, N. Clarke and J. W. Steed, Soft Matter, 2010, 6, 3541–3547 RSC.
-
(a) S.-I. Kawano, N. Fujita and S. Shinkai, J. Am. Chem. Soc., 2004, 126, 8592–8593 CrossRef CAS;
(b) J. Liu, J. Yan, X. Yuan, K. Liu, J. Peng and Y. Fang, J. Colloid Interface Sci., 2008, 318, 397–404 CrossRef CAS;
(c) A. Gasnier, G. Royal and P. Terech, Langmuir, 2009, 25, 8751–8762 CrossRef CAS.
-
(a) K. Haraguchi, Curr. Opin. Solid State Mater. Sci., 2007, 11, 47–54 CrossRef CAS;
(b) A. R. Hirst and D. K. Smith, Chem.–Eur. J., 2005, 11, 5496–5508 CrossRef CAS;
(c) A. Saha, S. Manna and A. K. Nandi, Chem. Commun., 2008, 3732–3734 RSC;
(d) D. Li, J. Liu, L. Chu, J. Liu and Z. Yang, Chem. Commun., 2012, 48, 6175–6177 RSC.
-
(a) C. R. Mayer, V. Cabuil, T. Lalot and R. Thouvenot, Angew. Chem., Int. Ed., 1999, 38, 3672–3675 CrossRef CAS;
(b) C. Wang, N. T. Flynn and R. Langer, Adv. Mater., 2004, 16, 1074–1079 CrossRef CAS;
(c) M. K. Shin, G. M. Spinks, S. R. Shin, S. I. Kim and S. J. Kim, Adv. Mater., 2009, 21, 1712–1715 CrossRef CAS;
(d) B. Tigges, C. Popescu and O. Weichold, Soft Matter, 2011, 7, 5391–5396 RSC;
(e) J. Y. Yook, G.-H. Choi and D. H. Suh, Chem. Commun., 2012, 48, 5001–5003 RSC;
(f) J. Wang, L. Lin, Q. Cheng and L. Jiang, Angew. Chem., Int. Ed., 2012, 51, 4676–4680 CrossRef CAS;
(g) Q. Wang, J. L. Mynar, M. Yoshida, E. Lee, M. Lee, K. Okuro, K. Kinbara and T. Aida, Nature, 2010, 463, 339–343 CrossRef CAS;
(h) Y. Zhou, N. Sharma, P. Deshmukh, R. K. Lakhman, M. Jain and R. M. Kasi, J. Am. Chem. Soc., 2012, 134, 1630–1641 CrossRef CAS.
-
(a) S. J. Rowan and J. B. Beck, Faraday Discuss., 2005, 128, 43–53 RSC;
(b) W. L. Leong, S. K. Batabyal, S. Kasapis and J. J. Vittal, Chem.–Eur. J., 2008, 14, 8822–8829 CrossRef CAS;
(c) J. Dash, A. J. Patil, R. N. Das, F. L. Dowdall and S. Mann, Soft Matter, 2011, 7, 8120–8126 RSC;
(d) T. D. Hamilton, D. K. Bucar, J. Baltrusaitis, D. R. Flanagan, Y. J. Li, S. Ghorai, A. V. Tivanski and L. R. MacGillivray, J. Am. Chem. Soc., 2011, 133, 3365–3371 CrossRef CAS;
(e) W. J. Gee and S. R. Batten, Chem. Commun., 2012, 48, 4830–4832 RSC;
(f) S. A. Joshi and N. D. Kulkarni, Chem. Commun., 2009, 2341–2343 RSC.
-
(a) M.-L. Cao, H.-G. Hao, W.-X. Zhang and B.-H. Ye, Inorg. Chem., 2008, 47, 8126–8133 CrossRef CAS;
(b) M.-L. Cao, H.-G. Hao and B.-H. Ye, Cryst. Growth Des., 2009, 9, 546–554 CrossRef CAS;
(c) M.-L. Cao, J.-J. Wu, H.-J. Mo and B.-H. Ye, J. Am. Chem. Soc., 2009, 131, 3458–3459 CrossRef CAS.
- M.-L. Cao, X.-L. Zhang and W. Yin, Chinese J. Inorg. Chem., 2011, 27, 1635–1641 CAS.
- J.-J. Wu, W. Xue, M.-L. Cao, Z.-P. Qiao and B.-H. Ye, CrystEngComm, 2011, 13, 5495–5501 RSC.
- A. Takahashi, M. Sakai and T. Kato, Polym. J., 1980, 12, 335–341 CrossRef CAS.
-
G. M. Sheldrick, SADABS 2.05, University of Göttingen, Germany, 2002 Search PubMed.
-
SHELXTL 6.10, Bruker Analytical Instrumentation, Madison, Wisconsin, USA, 2000 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- M. M. Smith and D. K. Smith, Soft Matter, 2011, 7, 4856–4860 RSC.
-
(a) Y. Jeong, M. K. Joo, Y. S. Sohn and B. Jeong, Adv. Mater., 2007, 19, 3947–3950 CrossRef CAS;
(b) K.-S. Moon, H.-J. Kim, E. Lee and M. Lee, Angew. Chem., Int. Ed., 2007, 46, 6807–6810 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: additional spectroscopic data for 3·NO3, 1, and 2. CCDC reference numbers 893054. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra22549b |
| ‡ The Tgel value is too high to measure when the concentration is lower than 2.8 wt%, and no phase transition was observed when the concentration is higher than 7.1 wt%. |
| § The freeze-drying process: one-year-aged gel or its solution was suddenly frozen by liquid nitrogen then dried in vacuum for 12 h. |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.