The high stability of intermediate radicals enhances the radical-scavenging activity of aminochromanols†
Keiko
Inami
*ab,
Ikuo
Nakanishi
c,
Mine
Morita
a,
Miyuki
Furukawa
a,
Kei
Ohkubo
d,
Shunichi
Fukuzumi
de and
Masataka
Mochizuki
ab
aKyoritsu University of Pharmacy, Shibako-en 1-5-30, Minato-ku, Tokyo 105-8512, Japan
bFaculty of Pharmaceutical Sciences, Tokyo University of Science, Yamazaki 2641, Noda-shi, Chiba 278-8510, Japan. E-mail: inami@rs.noda.tus.ac.jp
cResearch Center for Charged Particle Therapy, National Institute of Radiological Sciences (NIRS), Inage-ku, Chiba 263-8555, Japan
dDepartment of Material and Life Science, Graduate School of Engineering, Osaka University, ALCA, Japan Science and Technology Agency, Suita, Osaka 565-0871, Japan
eDepartment of Bioinspired Science, Ewha Womans University, Seoul 120-750, Korea
First published on 7th November 2012
Abstract
The hydroxyl radical-scavenging activity of the amino-substituted α-tocopherol analogues was much higher than that of α-tocopherol. Aminochromanoxyl radicals generated from 5- and 7-aminochromanol were successfully detected in aqueous solution at room temperature during ESR measurements. The stability of the aminochromanoxyl radicals leads to a significant enhancement of the radical-scavenging activity.
Reactive oxygen species (ROS) cause cancer, inflammation, and ischemia-reperfusion tissue damage and so on.1 Antioxidants, such as vitamin C and vitamin E prevent the oxidation of biomacromolecules.2–4 α-Tocopherol (1), a major component of vitamin E, is a very effective biological antioxidant that can scavenge peroxyl radicals in biological membranes, thus preventing oxidative damage.5 A large number of α-tocopherol analogues have been synthesized so far and assayed for their antioxidant activity and other types of physiological activities.6–10 The introduction of an electron-donating group at the C5, C7, or C8 position on the aromatic ring of α-tocopherol has been reported to elevate its antioxidant activity.4,9 2,2,5,7,8-Pentamethyl-6-chromanol (2) was often used as a model compound of 1, because the phytyl chain is not essential for the antioxidant activity in vitro.11 However, the structure–activity relationship for radical-scavenging reactions of α-tocopherol and its analogues has yet to be fully clarified. Herein, we report the synthesis of three α-tocopherol analogues (3–5) substituted with an amino group at the C5, C7, or C8 position of the aromatic ring (Fig. 1). The scavenging capacities of these compounds against hydroxyl radical (˙OH) and galvinoxyl radical (G˙) were also evaluated in order to shed light on the detailed structure–reactivity relationships of phenolic antioxidants.
 | ||
| Fig. 1 Structures of α-tocopherol and its analogues. | ||
2,2-Dimethyl-6-chromanol12 was nitrated at the C5 or C7 position on the chroman ring by NaNO213,14 or HNO3. The crude products were acetylated for purification, and the acetylated analogues were hydrolyzed after purification because of their unstable nature in the presence of light. The aminochromanols (3 and 4) were synthesized by the reduction of the corresponding nitrochromanols with Pd–C under H2 gas immediately before the antioxidant activity measurement. 8-Amino-6-chromanol (5) was synthesized by the hydrolysis of 6-acetoxy-8-amino-2,2-dimethylchroman, which was obtained by a simple catalytic reduction of the corresponding 6-acetoxy-8-nitro-2,2-dimethylchroman, which in turn was obtained by the nitration of 6-acetoxy-2,2-dimethylchroman with HNO3.
The ˙OH is an extremely reactive and short-lived ROS that is generated in the human body.1 The ˙OH-scavenging capacities of the compounds were estimated using the electron spin resonance (ESR) spin-trapping technique.15,16 The ESR assay was based on the competition between the reaction of ˙OH with a trapping agent and that with the synthesized compounds. The ˙OH was generated by the Fenton reaction, and 5,5-dimethyl-1-pyrroline N-oxide (DMPO) was used as the ˙OH-trapping agent.16 The capacity of the ˙OH scavenging activity at each antioxidant concentration was presented as a relative intensity, determined by calculating the ratio of the peak height of the ESR signal due to the ˙OH adduct of DMPO (DMPO–OH) to the peak height of the Mn2+ marker, which served as an internal standard. All of the amino compounds significantly inhibited the formation of DMPO–OH (Fig. 2). The ˙OH-scavenging capacity increased in the following order: 1–2 ≪ 5 < 3–4. In addition to the ESR signal due to DMPO–OH, another radical species derived from 3 and 4 was also detected around the g value of 2.004. The ESR signal intensity due to the unknown radical increased in a dose-dependent manner with respect to the addition of amino-substituted 6-chromanol (data not shown).
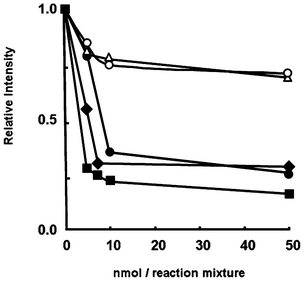 | ||
| Fig. 2 Hydroxyl radical scavenging activities of ESR for 1 (○), 2 (△), 3 (■), 4 (◆), and 5 (●). The reaction mixture was included with DMPO (0.20 M), 1–5 (0.02–1.14 × 10−3 M), FeSO4 (0.45 × 10−3 M), H2O2 (0.45 × 10−3 M) in a sodium phosphate buffer (pH 7.4) at 298 K. | ||
Since the ESR signals due to the unknown radical overlapped with those of DMPO–OH, G˙ was used instead of ˙OH to characterize the radicals in the absence of DMPO. When 3 or 4 was treated with G˙ in a phosphate buffer (pH 7.4) at room temperature, well-resolved ESR spectra were observed, indicating that hydrogen abstraction from the phenolic OH group in 3 or 4 by G˙ took place to produce the corresponding chromanoxyl radical, 3˙ or 4˙ (Fig. 3 or 4, respectively). The observed ESR spectra can be simulated with the hyperfine coupling constants (hfc) as shown in Fig. 3 and 4. The assignments of hfc values for 3˙ and 4˙ are carried out based on the isotropic Fermi contact couplings calculated by density functional theory (DFT) at the UB3LYP/6-31G(d) basis set as implemented in the Gaussian 09 program (Fig. 3c and 4c).17–19 The calculated values agree reasonably well with the experimental values determined from the ESR spectra. The hfc values demonstrate that the unpaired electron is highly delocalized over the nitrogen atom of the amino group. Such delocalization of the unpaired electron results in the enhanced stability of the aminochromanoxyl radicals.
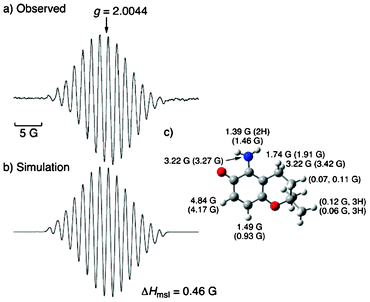 | ||
| Fig. 3 (a) ESR spectrum of 3˙ produced in the reaction of 3 (1.14 × 10−3 M) with G˙ (0.90 × 10−4 M) in a sodium phosphate buffer (pH 7.4) at 298 K. (b) Simulated spectrum of 3˙ using hfc and those predicted by DFT. (c) DFT optimized structure of 3˙ (UB3LYP/6-31(d) level) with hfc values together with the calculated values given in the parentheses. The maximum slope of the linewidths (ΔHmsl) value is shown below the spectra. | ||
 | ||
| Fig. 4 (a) ESR spectrum of 4˙ produced in the reaction of 4 (1.14 × 10−3 M) with G˙ (0.90 × 10−4 M) in a phosphate buffer (pH 7.4) at room temperature. (b) Simulated spectrum of 4˙ using hfc and those predicted by DFT. (c) DFT optimized structure of 4˙ (UB3LYP/6-31(d) level) with hfc values together with the calculated values given in the parentheses. The maximum slope of linewidths (ΔHmsl) value is shown below the spectra. | ||
The stability of the aminochromanoxyl radicals in aqueous solution was compared for the compounds with the amino group at the C5 or C7 position. G˙ alone did not decrease under these reaction conditions; however, the ESR signal intensity of 3˙ and 4˙ decreased in a time-dependent manner, as shown in Fig. 5. No differences in the decay rates of the intermediate radicals were observed between 3˙ and 4˙. The half-life of 3˙ and 4˙ in a phosphate buffer (pH 7.4) are about 2.64 × 103 s, which is about 340-fold larger than that for the α-tocopheroxyl radical (7.69 s) in ethanol.20 This indicates that the presence of the amino group at the ortho position relative to the oxyl radical is important for the stability of the aminochromanoxyl radicals.
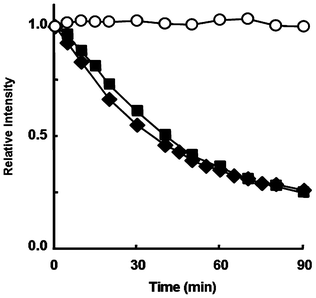 | ||
| Fig. 5 The stability of 3˙ (■), 4˙ (◆) and G˙ alone (○). The relative intensity of the ESR peak heights relative to Mn 2+. 3 or 4 (1.14 × 10−3 M) reacted with G˙ (0.90 × 10−4 M) in a sodium phosphate buffer (pH 7.4) at 298 K. | ||
The radical-scavenging activities of 1–5 were compared using second-order rate constants of the reaction with G˙.16,21 Upon addition of 2 to a deaerated acetonitrile solution of G˙, the absorption band at 428 nm due to G˙ disappeared precipitously. This behaviour indicates that hydrogen abstraction from the phenolic OH group in 2 by G˙ takes place, yielding the corresponding aminochromanoxyl radical and hydrogenated G˙.22,23 The decay in absorbance at 428 nm obeyed pseudo-first-order kinetics when the concentration of the compound was held at a greater than 10-fold excess relative to the G˙ concentration. The observed pseudo-first-order rate constant (kobs) increased linearly with the increasing concentration of the compound. From the linear plot of kobsvs. the concentration of the synthesized compounds, we determined the second-order rate constants (k) for hydrogen abstraction from 1 and 2 by G˙ to be 5.57 × 102 and 4.87 × 102 M−1 s−1, respectively. The k values for 3, 4 and 5 were determined in the same manner to be 1.81 × 103, 1.61 × 103 and 6.76 × 102 M−1 s−1, respectively. Thus, the k values for aminochromanols, especially 3 and 4, were much larger than those for 1 and 2.
All of the compounds synthesized in this study were found to react with ˙OH and G˙ efficiently. The aminochromanols 3 and 4, which have the amino group at the ortho position relative to the phenolic OH group, had higher radical scavenging activities than 1 and 2. A phenoxyl radical intermediate derived from 1 has been reported to be detected in organic solvent in the absence of oxygen by ESR. The fine structure has been determined, and it revealed that the unpaired electron was distributed mainly on the oxygen atom.24 In the reaction of the aminochromanols with ˙OH or G˙, intermediate radicals derived from 3 and 4 were well characterized as aminochromanoxyl radicals in aqueous solution at room temperature using ESR. The radicals formed were significantly stable at room temperature due to the delocalization of the unpaired electron over the nitrogen atom of the amino group. The introduction of an amino group on the chromanols did not result in differences in stability with respect to the substitution positions C5 or C7. The results showed that the electron-donating effect of the amino group on the chroman ring enhanced the radical-scavenging activity, as previously predicted,25 as a result of the higher stability of the radical intermediate.
Acknowledgements
This work was partially supported by Grant-in-Aid (Nos. 23590064, 23750014, and 20108010) from the Ministry of Education, Culture, Sports, Science and Technology, Japan, the Global COE (center of excellence) program “Global Education and Research Center for Bio-Environmental Chemistry” of Osaka University from MEXT, Japan and NRF/MEST of Korea through WCU (R31-2008-000-10010-0) and GRL (2010-00353) programs. We thank Dr T. Kawashima for his help in the computer simulations of the ESR spectra.References
- B. Halliwell and J. M. C. Gutteridge, Free radicals in biology and medicine, 3rd ed. Oxford University Press, Oxford, England, 1999 Search PubMed.
- E. Niki and N. Noguchi, Acc. Chem. Res., 2004, 37, 45 CrossRef CAS.
- C. K. Chow, Free Radical Biol. Med., 1991, 11, 215 CrossRef CAS.
- G. W. Burton and K. U. Ingold, Acc. Chem. Res., 1986, 19, 194 CrossRef CAS.
- S. Cuzzocrea, D. P. Riley, A. P. Caputi and D. Salvemini, Pharm. Rev., 2001, 53, 135 Search PubMed.
- (a) K. Mukai, Y. Kageyama, T. Ishida and K. Fukuda, J. Org. Chem., 1989, 54, 552 CrossRef CAS; (b) L. R. C. Barclay, M. R. Vinqvist, K. Mukai, S. Itoh and H. Morimoto, J. Org. Chem., 1993, 58, 7416 Search PubMed.
- (a) D. Shanks, R. Amorati, M. G. Fumo, G. F. Pedulli, L. Valgimigli and L. Engman, J. Org. Chem., 2006, 71, 1033 CrossRef CAS; (b) N. Al-Maharik, L. Engman, J. Malmström and C. H. Schiesser, J. Org. Chem., 2001, 66, 6286 CrossRef CAS.
- V. N. Povalishev, G. I. Polozov and O. I. Shadyro, Bioorg. Med. Chem. Lett., 2006, 16, 1236 Search PubMed.
- (a) G. W. Burton, T. Doba, E. J. Gabe, L. Hughes, F. L. Lee, L. Prasad and K. U. Ingold, J. Am. Chem. Soc., 1985, 107, 7053 CrossRef CAS; (b) W. Gregor, G. Grabner, C. Adelwöhrer, T. Rosenau and L. Gille, J. Org. Chem., 2005, 70, 3472 Search PubMed.
- (a) M. Wijtmans, D. A. Pratt, J. Brinkhorst, R. Serwa, L. Valgimigli, G. F. Pedulli and N. A. Porter, J. Org. Chem., 2004, 69, 9215 CrossRef CAS; (b) M. Wijtmans, D. A. Pratt, L. Valgimigli, G. A. DiLabio, G. F. Pedulli and N. A. Porter, Angew. Chem., Int. Ed., 2003, 42, 4370 CrossRef CAS.
- G. W. Burton and K. U. Ingold, J. Am. Chem. Soc., 1981, 103, 6472 CrossRef CAS.
- J. L. G. Nilsson, H. Sievertsson and H. Selander, Acta Chem. Scand., 1968, 22, 3160 CAS.
- A. Patel, F. Liebner, T. Netscher, K. Mereiter and T. Rosenau, J. Org. Chem., 2007, 72, 6504 Search PubMed.
- S. Yenes and A. Messeguer, Tetrahedron, 1999, 55, 14111 Search PubMed.
- G. R. Buettner, Free Radical Biol. Med., 1987, 3, 259 CrossRef CAS.
- D. Huang, B. Ou and R. L. Prior, J. Agric. Food Chem., 2005, 53, 1841 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. Montgomery, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford CT, 2009.
- (a) S. Fukuzumi, K. Ohkubo and T. Okamoto, J. Am. Chem. Soc., 2002, 124, 14147 CrossRef CAS; (b) K. Ohkubo, Y. Moro-oka and S. Fukuzumi, Org. Biomol. Chem., 2006, 4, 999 RSC; (c) S. E. Boesch and R. A. Wheeler, J. Phys. Chem. A, 1997, 101, 8351 CrossRef CAS.
- The coordinations and hfc values calculated by DFT are shown in Tables S1–S4 in ESI.†.
- K. Mukai, A. Ouchi, A. Mitarai, K. Ohara and C. Matsuoka, Bull. Chem. Soc. Jpn., 2009, 82, 494 Search PubMed.
- N. Gotoh, K. Shimizu, E. Komuro, J. Tsuchiya, N. Noguchi and E. Niki, Biochim. Biophys. Acta, Lipids Lipid Metab., 1992, 1128, 147 Search PubMed.
- (a) I. Nakanishi, T. Kawashima, K. Ohkubo, H. Kanazawa, K. Inami, M. Mochizuki, K. Fukuhara, H. Okuda, T. Ozawa, S. Itoh, S. Fukuzumi and N. Ikota, Org. Biomol. Chem., 2005, 3, 626 RSC; (b) I. Nakanishi, K. Fukuhara, T. Shimada, K. Ohkubo, Y. Iizuka, K. Inami, M. Mochizuki, S. Urano, S. Itoh, N. Miyata and S. Fukuzumi, J. Chem. Soc., Perkin Trans. 2, 2002, 1520 RSC.
- L. Valgimigli, K. U. Ingold and J. Lusztyk, J. Am. Chem. Soc., 1996, 118, 3545 CrossRef CAS.
- (a) M. Matsuo and S. Matsumoto, Lipids, 1983, 18, 81 Search PubMed; (b) J. Tsuchiya, E. Niki and Y. Kamiya, Bull. Chem. Soc. Jpn., 1983, 56, 229 CAS; (c) T. Ozawa and A. Hanaki, Biochem. Biophys. Res. Commun., 1985, 126, 873 Search PubMed.
- (a) J. S. Wright, E. R. Johnson and G. A. DiLabil, J. Am. Chem. Soc., 2001, 123, 1173 CrossRef CAS; (b) J. S. Wright, D. J. Carpenter, D. J. McKay and K. U. Ingold, J. Am. Chem. Soc., 1997, 119, 4245 CrossRef CAS; (c) W. Chen, P. Guo, J. Song, W. Cao and J. Bian, Bioorg. Med. Chem., 2006, 12, 3582 Search PubMed; (d) Y. K. Tyagi, A. Kumar, H. G. Raj, P. Vohra, G. Gupta, R. Kumari, P. Kumari and R. K. Gupta, Eur. J. Med. Chem., 2005, 40, 413 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: synthesis and characterization of 3–5. See DOI: 10.1039/c2ra21928j |
| This journal is © The Royal Society of Chemistry 2012 |