Ring-opening polymerization of lactones using supramolecular organocatalysts under simple conditions†
Coralie
Thomas
ab,
Frédéric
Peruch
*b and
Brigitte
Bibal
*a
aUniversité de Bordeaux, Institut des Sciences Moléculaires, UMR CNRS 5255, 351 cours de la Libération, 33405 Talence, France. E-mail: b.bibal@ism.u-bordeaux1.fr; Fax: 33 54000 6158; Tel: 33 54000 3364
bUniversité de Bordeaux, LCPO, UMR 5629, F-33600 Pessac, France and CNRS, LCPO, UMR 5629, F-33600 Pessac, France. E-mail: peruch@enscbp.fr; Fax: 33 54000 8487; Tel: 33 54000 2745
First published on 19th October 2012
Abstract
Ring-opening polymerizations of δ-valerolactone (δ-VL) and ε-caprolactone (ε-CL) were catalyzed by a metal-free system composed of two H-bonding components, a phenol derivative to activate the monomer, and DBU, which enhanced the nucleophilicity of the initiator and the propagating chain. Compared to other H-bonding systems for the ROP of lactones, phenol + DBU catalysts had the practical advantages of being commercially available and inexpensive, efficient at room temperature and under simple experimental conditions that avoid drying of reactants and the use of a glove-box. In addition, the obtained polyesters had a narrow dispersion of molar masses which were controlled by the concentration ratio of monomer versus initiator. Moreover, the initiation of the polymerization by DBU and residual water molecules (despite no specific drying of reagents) was shown to be very minor under the experimental conditions. No initiation by the phenol catalysts was observed. Block copolyesters PVL-PLA and PCL-PLA were also prepared using these conditions.
Introduction
Organocatalyzed ring-opening polymerization (ROP) has gained attention since 2001 and the first report of living ROP of lactide catalyzed by DMAP, in the presence of a primary alcohol as an initiator.1 The main advantage of this catalysis is that it consists of a metal-free process that allows preparation of polymers for further applications in biomedical materials and food packaging.2 Over the last decade, a broad range of organocatalysts were demonstrated to be efficient in the preparation of polylactides, based on the activation of the reactant(s) (monomer, initiator, polymer growing chain), through hydrogen bonds (H-bonds) and/or temporary covalent bonds. So, strong bases (amidine, guanidine, phosphazene), H-bond donors (thioureas, amides, sulfonamides, amino-thiazolines, fluorinated alcohols), Brönsted acids, and nucleophilic compounds (phosphines, N-heterocyclic carbenes) catalyzed the ROP of lactide in a reasonable reaction time (2–48 h) with a moderate loading (1–10 mol%).3,4 Generally, reactions were conducted at 20–40 °C, under drastic anhydrous conditions that required a careful drying of the solvent, the reactants and the catalysts, as well as the use of a glove-box. Polylactides had narrow dispersity (D ∼ 1.10–1.30) and the molar masses depended on the ratio of monomer versus initiator. At present, research interests have shifted towards a more sustainable ROP of lactide5 and the ROP of other cyclic monomers,3 such as cyclic carbonates, lactones, epoxides and carbosiloxanes.Among the organocatalytic systems designed for ROP, H-bonding ones appeared to be highly controlling the polymerization through a smooth living-like process, and thus allowing the preparation of very narrowly dispersed polymers, with controlled masses. In 24 h, the conversions can reach 90–100% for lactide and δ-valerolactone. When conversion of monomers is close to 100%, waste of starting material is minimal and purification of the polymeric product is easier. H-bonding catalysts also present some limitations in terms of activation: most polymerize lactide, but few were described for the preparation of polyesters from lactones. For example, TBD was reported as a bifunctional catalyst for the ROP of δ-valerolactone (δ-VL) and ε-caprolactone (ε-CL) in d6-benzene (Scheme 1).6 Dual systems based on thiourea TU in partnership with DBU or MTBD also proved to be efficient under the same conditions: high monomer conversion (92–95% in 3–4 h for δ-VL, 78% in 5d for ε-CL), controlled molar masses of the polyesters in the 4000–17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 range and low dispersity (D = 1.04–1.06).6 Nevertheless, the disadvantages of the former systems were the drastic experimental conditions (careful drying of reactants, use of a glove box) and the cost of TU synthesis.
000 g mol−1 range and low dispersity (D = 1.04–1.06).6 Nevertheless, the disadvantages of the former systems were the drastic experimental conditions (careful drying of reactants, use of a glove box) and the cost of TU synthesis.
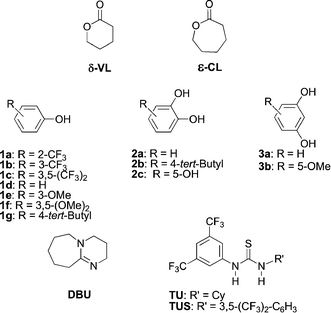 | ||
| Scheme 1 δ-Valerolactone (δ-VL) and ε-caprolactone (ε-CL), Hydrogen-bonding (donor + acceptor) catalysts. | ||
In this context, the development of new organocatalysts for the ROP of lactones is highly desirable, aiming at a minimum cost for catalysts, a simple protocol running at room temperature, and optimum catalytic properties that induce a full conversion (no waste material) and a controlled polymer structure. In addition, if possible, the reaction time should be minimized (24 h maximum). Recently, we reported the controlled ROP of lactide in dichloromethane, using a commercially available catalytic system, that was efficient under simple experimental conditions avoiding glove box use. It was made up of a H-bond donor (phenols provided with electron-withdrawing groups) and a H-bond acceptor (tertiary amine such as CyNMe2 and (–)-sparteine), in the presence of 4-biphenylmethanol (BPM), as an initiator.7 Polylactides with predictable masses between 3100 and 8400 g mol−1 and low dispersity (D = 1.03–1.08) were obtained.
Herein, under simple experimental conditions (20 °C, no drying of reactants), we investigated and optimized the simple catalytic system, phenol 1–3 + DBU in the ROP of model lactones, δ-VL and ε-CL (Scheme 1). These phenol-based systems were also compared to thiourea-based ones, using TU and the Schreiner's catalyst TUS. The extension of the methodology to block copolymers was then exploited.
Results and discussion
Mechanistic discussion
A main challenge in organocatalysis is the determination of the reaction mechanism that usually involves transient species and/or complex intermediates. In the case of the ROP of δ-VL and ε-CL, catalyzed by a dual H-bonding system and initiated by a primary alcohol, the classical mechanism relies on the formation of two different H-bonded complexes between the partner catalysts and the reactants (Scheme 2a):6,7 a (phenol:δ-VL) complex and a (H-bond acceptor catalyst (DBU):alcohol) complex, with R′OH alcohol representing the initiator (BPM) and the growing chain. These H-bonds induce an increased reactivity of the monomer and the growing chain, that allows the extension of the polymer chain. To further support this supramolecular mechanism, titrations monitored by 1H NMR (CDCl3) were conducted to characterize H-bonds in a model system for the ROP of δ-VL:phenol 1d as a H-bond donor, DBU as H-bond acceptor and BPM as the initiator. An association constant of 63 M−1 (K2, Scheme 2a) was determined between DBU and BPM, whereas no obvious binding (K1 ∼ 0) between phenol 1d and δ-VL was observed, presumably due its weakness. Concerning the other possible H-bonds in the reaction medium (i.e. phenol with its co-catalyst), we already showed that undesired interactions between the catalysts could decrease their activity. But, depending on the overall equilibrium, the latter complexes could be sufficiently displaced towards those that induced the polymer synthesis. So, in the proposed model, we found that the non-desired interactions between the two catalysts were also present: a binding constant of 85 M−1 was found between phenol 1d and DBU. As previously demonstrated, even in the presence of undesired H-bonds, weak H-bonds involved in the ROP mechanism (K1–2 ≤ 2 M−1) could trigger polymerization.8
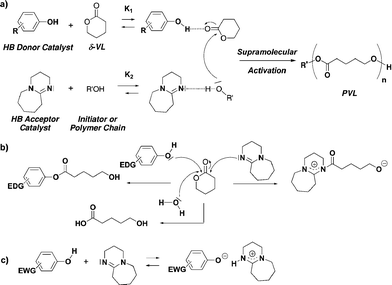 | ||
| Scheme 2 a) Proposed mechanism for the ROP of lactones catalyzed by H-bonding systems (phenol 1–3 + DBU) in presence of an alcohol initiator. b) Possible nucleophilic attack of catalysts and a water molecule on monomer. c) Potential acido-basic reaction between catalysts. | ||
Other mechanistic possibilities should also be discussed, as the chosen catalysts could display different properties depending on experimental conditions: H-bonding, nucleophilic and acido-basic ones. So the expected supramolecular mode of activation could be in competition with (i) a nucleophilic attack of electron-rich phenols, water and amines/amidines/guanidines on the monomer (Scheme 2b), and (ii) an acido-basic reaction between phenols and amines/amidines/guanidines (Scheme 2c). These side reactions could allow the occurrence of reactive alcoholate or alcohol species in the medium, that could lead to polymer chains ended by different groups (phenol ester, carboxylic acid, amidium), to the detriment of a controlled mechanism.
Concerning possible nucleophilic attacks on the monomer (Scheme 2b), our group showed that phenols provided with electron–donating groups could sometimes initiate the ROP of lactide, due to the high reactivity of this monomer towards nucleophilic attack.7 However, lactones were less electrophilic than lactide, and they should be less reactive towards phenols. Mass spectrometry analysis of the polyesters will be duly done to highlight potential side-initiation by phenols. Based on a similar reactivity, a water molecule could also initiate the polymerization (Scheme 2b). As our experimental conditions included no drying of the reactants, this potential side-reaction could also be observable by mass spectrometry analysis of the polymers. Besides, the nucleophilicity of the selected Lewis base DBU was known as nucleophilic and a poor leaving group, possibly leading to stable amidiniums.9 So, experimental conditions should be tuned to avoid undesired nucleophilic reactions between the co-catalyst and the monomer, using an appropriate ratio of monomer vs. amine as well as a H-bond acceptor (initiator, polymer chain) that duly interact with the amine. Mass spectrometry analysis of the polyesters will again give indication on the occurrence of this nucleophilic side-reaction.
With regards to the hypothetical acido-basic reaction between the two catalysts, we checked that their pKA values in acetonitrile were not compatible with a proton exchange: pKA CH3CN (phenols) = 26–2810 and pKA CH3CN (DBU) = 24.3.11 Making the assumption that the value of their difference in pKA in acetonitrile (superior to 2 units) is close to the one in dichloromethane, phenolate species should be produced in a very small amount during the ROP reaction, avoiding an anionic propagation mechanism. This assumption will be checked by analyzing the polyesters end-group by mass spectrometry.
In conclusion, keeping in mind that undesired interactions and/or reactions were plausible, it is important to evaluate the activity of the chosen catalysts, i.e. H-bonding vs. potential nucleophilic initiation by these species. To the best of our knowledge, this is the first time that two nucleophilic H-bonding organocatalysts were evaluated for the ROP of cyclic esters.
Dual catalytic system phenol + DBU in the ROP of lactones
After a preliminary optimization of the best H-bond acceptor among (–)-sparteine, BTMG, DBN, DBU and MTBD, we found that DBU gave the highest conversion of δ-VL in presence of phenols as H-bond donor catalyst (see ESI†, Table S1) and a primary alcohol as an initiator. The ROP reactions of δ-VL (4 M) were achieved in dry dichloromethane at 20 °C, in the presence of the catalytic system DBU + H-bonding catalyst 1–3 (5 mol% each, with respect to the monomer) and 4-biphenylmethanol (5 mol%) as an initiator, 4 Å molecular sieves (Table 1) without pre-drying of the reactants.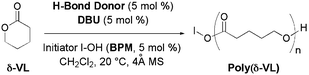
| Run | Catalysts | BPM (mol %) | Conv. (%) | Mn THEOc (g mol−1) | Mn SECd (g mol−1) | De |
|---|---|---|---|---|---|---|
| a Conditions of ROP: Lactone (4 M in dichloromethane), H-bond donor catalyst (5 mol %), DBU co-catalyst (5 mol %), 4-biphenylmethanol as an initiator (5 mol %), 4 Å molecular sieves, 20 °C, 24 h. b Polylactone characterized by SEC when conversion was higher than 75%. c Theoretical molar mass in g mol−1 related to conversion. d Experimental molar masses determined by size exclusion chromatography (SEC). e Dispersity determined by SEC. f n.d.: not determined. g Sequential chain extension by adding the same quantity of monomer at t = 0, 24 h, 48 h and stirring for 72 h. | ||||||
| 1 | DBU | 0 | 25 | n.d.f | n.d.f | n.d. |
| 2 | DBU | 1 | 42 | 4780 | 3340 | 1.18 |
| 3 | DBU | 5 | 44 | 1060 | n.d. | n.d. |
| 4 | DBU + TU | 5 | 97 | 2110 | 2920 | 1.15 |
| 5 | DBU + TU | 1 | 86 | 8760 | 5640 | 1.08 |
| 6 | DBU + TUS | 5 | 91 | 1990 | 2250 | 1.17 |
| 7 | DBU + TUS | 1 | 89 | 9060 | 6890 | 1.08 |
| 8 | DBU + 1a | 5 | 82 | 1790 | 1230 | 1.10 |
| 9 | DBU + 1a | 1 | 40 | 4070 | 2750 | 1.09 |
| 10 | DBU + 1b | 5 | 71 | 1600 | n.d. | n.d. |
| 11 | DBU + 1c | 5 | 65 | 1480 | n.d. | n.d. |
| 12 | DBU + 1d | 5 | 94 | 2050 | 1780 | 1.07 |
| 13 | DBU + 1d | 1 | 75 | 7640 | 4325 | 1.08 |
| 14 | DBU + 1e | 5 | 91 | 1990 | 2380 | 1.13 |
| 15 | DBU + 1e | 1 | 75 | 7640 | 3900 | 1.14 |
| 16 | DBU + 1f | 5 | 94 | 2050 | 1700 | 1.07 |
| 17 | DBU + 1f | 1 | 76 | 7750 | 3840 | 1.15 |
| 18 | DBU + 1g | 5 | 91 | 1990 | 2010 | 1.09 |
| 19 | DBU + 1g | 1 | 76 | 7740 | 4210 | 1.12 |
| 20 | DBU + 1g | 5g | 86, 100, 91 | 1880, 3880, 5700 | 1920, 4230, 5860 | 1.08, 1.11, 1.13 |
| 21 | DBU + 2a | 5 | 97 | 2090 | 2200 | 1.10 |
| 22 | DBU + 2a | 1 | 84 | 8550 | 5840 | 1.11 |
| 23 | DBU + 2b | 5 | 97 | 2120 | 2360 | 1.09 |
| 24 | DBU + 2b | 1 | 90 | 9160 | 5510 | 1.07 |
| 25 | DBU + 2b | 5g | 100, 67, 38 | 2180, 3520, 4280 | 2260, 2870, 2960 | 1.11, 1.14, 1.12 |
| 26 | DBU + 2c | 5 | 96 | 2090 | 1670 | 1.10 |
| 27 | DBU + 2c | 1 | 68 | 6920 | 4200 | 1.05 |
| 28 | DBU + 2c | 5g | 98, 62, 47 | 2140, 3380, 4320 | 2240, 2850, 3150 | 1.13, 1.12, 1.10 |
| 29 | DBU + 3a | 5 | 98 | 2140 | 1410 | 1.08 |
| 30 | DBU + 3a | 1 | 46 | 4680 | 3360 | 1.08 |
| 31 | DBU + 3b | 5 | 93 | 2120 | 1620 | 1.10 |
| 32 | DBU + 3b | 1 | 42 | 4280 | 3540 | 1.06 |
The conversion of δ-VL was remarkable (90–100%) when DBU was combined with TU, TUS and electron-rich phenols 1d–g, catechols 2a–b, pyrogallol 2c, resorcinols 3a–b. The moderate results obtained with phenols containing electron-withdrawing groups 1a–c (65–82% conv.) were attributed to their stronger H-bond with DBU. The resulting crude polymers were analyzed by size exclusion chromatography (SEC) using refractive index (RI) and UV detectors with standard polystyrene calibration and/or triple detection (RI, viscosimetry and light scattering). For such low molar masses, it was shown that no correction factor has to be applied, which was usually the case for polylactones molar masses measured by SEC with polystyrene calibration, as molar masses estimated with all type of detection were very close. Experimental molar masses (MnSEC) were in fairly good agreement with the theoretical values (MnTHEO) (see ESI†, Fig. S15–17) and the dispersity of the polyesters was fairly narrow (D ≤ 1.15).
As previously demonstrated by us,7 both SEC and mass spectrometry analyses were necessary for evaluating the degree of control for the polymerization reactions, even with a low dispersity. Mass spectra indicated that these polyesters were chain-ended by the initiator and, in some cases, polymers chain-ended by water were seen (see ESI†, Fig. S1), as we directly used commercial compounds for easy technical purposes (no reactant drying, no glove-box). Of note, no initiation by the phenol catalysts under these conditions was noticed, contrary to what we observed in the ROP of lactide catalyzed by phenol/Sp/BPM in 5:5:5 ratio.
Indeed, several experimental results indicated that polymer initiation by water molecules was minor. First, mass spectra (ESI†, Fig. S1–S2) clearly indicated that the chains ended by water were shorter than those ended by BPM. As the intensity of peaks in MALDI-TOF was not representative of the actual family concentration, other analyses have to be achieved to evaluate the amount of water initiation. Secondly, comparing experimental masses detected by UV (only chains ended by BPM were seen) and refractometric index (polymer chains ended by any initiator were detected), the values were very close, confirming that the major polymer was chain-ended by BPM. Thirdly, as 1H NMR spectra were not accurate (see ESI†, Fig. S5), 13C NMR spectra of the crude polymers were inspected in the carbonyl region to discriminate the possible different end-groups: benzylic ester (BPM as an initiator) and carboxylic acid (water as an initiator). In the 170–180 ppm region, the signals of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O from polyester (δ = 174.2 ppm) were observed and a tiny signal of carboxylic acid (δ = 177.6 ppm) was noticed (ESI†, Fig. S4). The latter signal could correspond to the excess of benzoic acid, introduced as a DBU quencher, and to an acid chain-end. So, even if the acid signal was assigned to pure initiation of polymerization by water, this phenomenon was detected by 13C NMR as weak. Combining these results, the water initiation can be considered to be a minor phenomenon in the proposed protocol.
O from polyester (δ = 174.2 ppm) were observed and a tiny signal of carboxylic acid (δ = 177.6 ppm) was noticed (ESI†, Fig. S4). The latter signal could correspond to the excess of benzoic acid, introduced as a DBU quencher, and to an acid chain-end. So, even if the acid signal was assigned to pure initiation of polymerization by water, this phenomenon was detected by 13C NMR as weak. Combining these results, the water initiation can be considered to be a minor phenomenon in the proposed protocol.
ROP of δ-VL was also performed in the presence of 1 mol% of initiator, using the most efficient catalytic systems (Table 1). In 24 h, the best conversions (≥85%) were observed when DBU was used in partnership with thioureas or catechols 2a–b. Narrowly dispersed polyesters (D = 1.08–1.11) were obtained with masses in poor agreement with the theoretical ones. The mass spectra showed that the polymers were initiated by BPM, water and DBU (ESI, S2†). Thus, the nucleophilic character of DBU could be expressed under a lower loading of initiator, and it overtook over its H-bonding abilities. Considering the H-bonding mechanism (Scheme 2a), it could be easily explained that by decreasing the BPM/DBU ratio, uncomplexed DBU could interact/react with different species present in the medium (water, monomer, catalyst), and thus unbalance the whole catalytic system and the outcome of the reaction. Obviously, the proposed catalytic systems at 5 mol% were more efficient in the presence of 5 mol% BPM, and led to perfectly controlled polyvalerolactones under optimum conditions, phenol:DBU:BMP in 5/5/5 ratio related to the monomer.
Thus, to increase molar masses of polyesters without unbalancing the H-bonding mechanism, chain elongation experiments consisting in successive additions of monomer, were performed in the presence of phenol:DBU:BMP in 5/5/5 ratio. In partnership with DBU, three efficient catalysts phenol 1g, catechol 2b and pyrogallol 2c were tested and, for each one, three independent experiments were conducted during 24 h, 48 h and 72 h, with reloading the same quantity of δ-VL after 24 h and 48 h. In terms of efficiency, the best system was 1g + DBU, that yielded to an excellent conversion at each step (86–100%, Table 1). Molar masses of polyesters were in good correlation with the predicted masses at 24 h, 48 h and 72 h whereas dispersity was fairly narrow (D = 1.08–1.13). Sequential polymerizations conducted with 2b + DBU and 2c + DBU were slower at each step (from ∼100% after 24 h to ∼40–50% conversion at the final step). The SEC analyses of the corresponding polylactones indicated lower masses than expected. So, catechol or pyrogallol + DBU systems underwent less controlled and less efficient reactions over extended periods (>24 h), probably due to the decreased amount of DBU available in the medium as a catalyst and increased quantity of DBU as chain-end of polymers. This phenomenon could be attributed to a disequilibrium in the H-bonding mechanism (Scheme 2a–b) towards the nucleophilic attack of DBU on the monomer. It could be proposed that H-bonds between DBU and catechol/resorcinol were weaker than with phenol 1g, pre-empted by an intramolecular H-bond. Then, in these 72 h experiments, DBU showed dual properties, nucleophilic and H-bonding ones. Thus the best catalytic system for the ROP of δ-VL was 1g + DBU (5 mol%) in presence of BPM 5 mol%, allowing the preparation of polyvalerolactone with masses of 2000–5700 g mol−1, through a living-like process (as shown by the increase of molar masses with monomer addition).
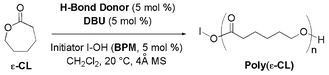
The reactivity of ε-CL in presence of H-bonding catalysts was known to be lower than the one of δ-lactones.6 The organocatalyzed ROP of ε-CL was thus investigated under the same conditions (Table 2) but for extended periods (48 h and 120 h). At first, ROP were conducted in the presence of DBU, without (not shown in Table 2) and with BPM. After 120 h, DBU alone had no impact on the monomer (i.e. 0% conversion) whereas 11% of poly-ε-caprolactone (PCL) was observed when DBU was employed in the presence of BPM (5 mol%).
| H-Bond donor | None | TU | TUS | 1a–c | 1d | |
|---|---|---|---|---|---|---|
| a Conditions of ROP: ε-caprolactone (4 M in dichloromethane), H-bond donor catalyst (5 mol %), DBU co-catalyst (5 mol %), 4-biphenylmethanol as an initiator (5 mol %), 4 Å molecular sieves, 20 °C, 48 h or 120 h. | ||||||
| Conv. (%) | 48 h | n.d. | 37 | 1 | 0–8 | 17 |
| 120 h | 11 | 45 | 5 | 14–27 | 35 | |
| H-Bond donor | 1e–g | 2a | 2b | 2c | 3a–b | |
| Conv. (%) | 48 h | 4–18 | 29 | 53 | 38 | 12 |
| 120 h | 35–39 | 66 | 80 | 57 | 19–22 | |
ROP of ε-CL catalyzed by H-bond donor + DBU systems was moderately efficient in 48 h: thiourea TUS, phenols 1a–g and resorcinols 3a–b, had a poor conversion (0–18%) whereas thiourea TU and catechols 2a–c had a 29–53% conversion. The latter systems were then tested over 120 h. Indeed, the reaction progress was then improved. Notably, catechols 2a–c + DBU catalysts induced 57–79% conversion of ε-CL. These results were similar to the other H-bonding catalytic systems in d6-benzene.6 In the presence of TU + DBU systems (Table 2), only 45% of PCL was obtained under the same period. From mass analysis, these polylactones were only chain-ended by the initiator (ESI†, Fig. S3). This observation was also supported by the 13C NMR spectra of the crude PCL (see ESI†, Fig. S6–7: no detectable carboxylic acid group as a terminal chain-end).
Copolymerizations
Due to complementary properties of both PLA and PCL (biodegradability, mechanical properties), block copolymers of LA and ε-CL are particularly of keen interest leading to a wide range of biomedical devices with adjustable properties depending on the composition, monomer sequencing and molecular weights. Our methodology was thus implemented to prepare block copolymers composed of DL-lactide (LA), δ-VL and ε-CL (Table 3) by sequential polymerization using a selection of catalytic systems (DBU in partnership with phenol 1d, o-CF3-phenol 1a, 4-tert-butyl-catechol 2b, pyrogallol 2c or resorcinol 3a). The first block (block A) and the final polymers (AB) were synthesized under our experimental conditions and analyzed by 1H and 13C NMR, SEC and differential scanning calorimetry (DSC).| Successive monomers | Catalytic system | Conv. (%)c | MnSEC A, MnSEC AB (MnTHEO) (g mol−1)d | DA, ABe |
|---|---|---|---|---|
| a Conditions of ROP: Monomer A (lactone 4 M in CH2Cl2) then monomer B (solid lactide), H-bond donor catalyst (5 mol %), DBU co-catalyst (5 mol %), propan-1-ol as an initiator (5% mol), 4 Å molecular sieves, 20 °C. b Reaction times 24 h for Lactide and δ-VL; 120 h for ε-CL. c Percentage of conversion regarding monomers A and B respectively determined by 1H NMR (CDCl3). d Theoretical molar mass in bracket and experimental ones determined by size exclusion chromatography (SEC); A after completion of the first step, AB after completion of the second step. e Dispersity determined by SEC. | ||||
| δ-VL, LA | DBU + 1a | 87, 100 | 2680, 5550 (1790, 4670) | 1.15, 1.32 |
| DBU + 1d | 96, 100 | 2800, 5010 (1980, 4860) | 1.18, 1.35 | |
| DBU + 2b | 96, 100 | 2820, 4930 (1980, 4860) | 1.25, 1.30 | |
| DBU + 2c | 96, 100 | 2590, 4950 (1980, 4860) | 1.23, 1.32 | |
| DBU + 3a | 93, 100 | 2710, 5210 (1910, 4790) | 1.15, 1.39 | |
| ε-CL, LA | DBU + 1a | 21, 100 | 1620, 3950 (500, 2500) | 1.08, 1.48 |
| DBU + 1d | 40, 100 | 3350, 3970 (940, 2940) | 1.04, 1.25 | |
| DBU + 2b | 95, 100 | 3950, 6050 (2220, 4220) | 1.15, 1.22 | |
| DBU + 2c | 74, 100 | 2790, 5640 (1730, 3730) | 1.09, 1.26 | |
| DBU + 3a | 30, 100 | 1520, 3910 (700, 2700) | 1.08, 1.38 | |
The polyvalerolactone-polylactide (PVL-PLA) polymers were obtained in 48 h with a high overall percentage of conversion (87–100%). By 1H NMR monitoring, we checked that the unreacted VL after 24 h, was not incorporated during the following 24 h. Each monomer was converted at the same level as when the experiments were carried out independently (Table 1). The experimental molar masses of block A (MnA) and the copolymer (MnAB) from RI and UV detection (see ESI†, Fig S20–22) were close to the theoretical values, as expected when the process of polymerization was living-like at each synthetic step, and thus controlled by the ratio [consumed monomer]/[initiator]. The dispersities of block A and copolymer were fairly narrow (D = 1.15–1.39). On the 1H NMR spectra of the polyesters, the absence of PVL chain-ends (–CH2–OH signal at 3.6 ppm) was also a proof of the quantitative initiation of lactide polymerization by the PVL macroinitiator (see ESI†, Fig. S9–10). The 13C NMR of the PVL-PLA copolymers clearly indicated the presence of only two carbonyl signals, at δ = 169.3 & 169.6 and 173.0 ppm corresponding to the PLA block (two signal due to DL-LA reactant, without transesterification reaction) and the PVL block, respectively (ESI†, Fig. S8). The DSC analysis of the copolymers showed a glass transition (Tg) at −43 °C, a crystalline point (Tc) at 13 °C and a melting point (Tm) at 28 °C, which were a little bit lower than data from the corresponding homopolymers of δ-VL (Tg = −63 °C, Tm = 60 °C) and DL–LA (Tg = 45–50 °C), as expected for measurements achieved on oligomers (ESI†, Fig. S13).12
Then, sequential organocatalyzed ROP of ε-CL then LA were achieved in 6 days. Block A incorporated 21 to 95% of ε-CL and block B was made of the total quantity of LA. 1H NMR spectra indicated that the residual CL after 120 h, was not incorporated during the following 24 h. Conversion of the first monomer was in the same order as expected (15–79%, Table 1). Molar masses of block A and copolymer (ESI†, Fig. S22–24) were smaller than the theoretical values, however, in most cases dispersity was low (1.04–1.26, except the two copolymers obtained by DBU + 1a and DBU + 3a). On the 1H NMR spectra (ESI†, Fig. S12), PLA chain-ends (CHMe–OH) at 4.4 ppm were seen and PCL terminal group was not present. The analysis of the copolymers by 13C NMR clearly indicated that they were made of two homo-dyad sequences CL–CL and LA-LA (ESI†, Fig. S11). Indeed, even if the polymerization of the first block (CL) was generally far from being complete, the second block was only constituted of LA units, because of the high reactivity difference between the two blocks (few hours compared to several days). The DSC thermogram of the copolyesters obtained from DBU + 2b catalysis (allowing higher CL conversion) displayed temperatures Tg at −49 °C, Tm at 43 °C (Tc was observed at −3 °C) that were lower than those of PCL (Tg = −60 °C, Tm = 65 °C) and DL-PLA (ESI†, Fig. S14).12 Again, the observed differences could be explained by the low masses of the polyesters.
Thus, block copolymers PVL-PLA were synthesised using several (phenol + DBU) catalysts, in a controlled manner with high conversion. Notably, the block PCL-PLA copolymer was obtained with DBU + 2b system with a high conversion for H-bonding catalysis.
Conclusions
The ROP of δ-VL organocatalyzed by dual H-bonding systems based on phenol derivatives, was optimized using DBU as an activator of the nucleophilic species in the medium. The polymerization process was efficient (high conversion in 24 h) and controlled (MnSEC ∼ MnTHEO and D ≤ 1.13) through a supramolecular mechanism. Under these conditions, ε-CL was polymerized in 120 h in a controlled manner and the 4-tert-butyl-catechol 2b + DBU system underwent the highest conversion (80%). The methodology was successfully extended to block copolymers PVL-PLA and PCL-PLA. Moreover, the mechanistic pathway involving the nucleophilic attack of the monomer by DBU and residual water molecules (despite no specific drying of reagents) was shown to be very minor under the experimental conditions. No initiation by the phenol catalysts was observed.Compared to the known H-bonding systems described for the ROP of lactones,5 phenol and DBU are commercially available, and inexpensive, used without any purification and are highly efficient under classical bench conditions (no glove-box is required). These practical advantages are crucial for the development of organocatalyzed ring-opening polymerization of cyclic esters, accessible for any chemist.
Experimental
Materials
Dichloromethane was dried over calcium hydride and distilled. Thioureas TU and TUS were prepared according to the literature.13 Commercially available phenols 1–3, (–)-sparteine, BTMG, DBN, DBU and MTBD as well as lactones were used as received.General procedure for ring-opening polymerization of lactones
Under nitrogen, in a dry Schlenk tube, were successively introduced dry dichloromethane (0.5 mL), lactone (1 or 2 mmol), phenol catalyst 1–3 (5 mol %), the amine co-catalyst (5 mol %), 4-biphenylmethanol (1 or 5 mol %), and 4 Å molecular sieves (5 beads). The reaction mixture was stirred at 20 °C under nitrogen for the indicated period. The reaction mixture was quenched with benzoic acid (10 mg), filtered and concentrated in vacuo. Conversion was determined by 1H NMR, integrating the signals of the methine proton (adjacent to the carbonyl group) in both the residual lactone and the polymer. Polymer molar masses and dispersity were determined by size exclusion chromatography (SEC) on apparatus equipped with Tosoh GSK4000, GSK3000 and GSK2000 columns (Eluent: THF, flow rate 1.0 mL/min, 40 °C), either with RI and UV detectors and a calibration with polystyrene standards, or with triple detection (RI, Light Scattering and viscosimetry) with universal calibration (dn/dc = 0.079 mL/g for PCL). DSC measurements were carried out using a Perkin Elmer DSC7 instrument calibrated with cyclohexane. For the successive heating/cooling cycling measurements, the following thermal procedure was used: isotherm at −90 °C for 1 min, 10 °C/min ramp from −90 °C to 200 °C, isotherm at 200 °C for 1 min and 10 °C/min ramp from 200 °C to −90 °C.General procedure for copolymerization
The previous procedure was applied for the preparation of block A using the appropriate monomer (δ-VL or ε-CL at 4M). After the indicated period, monomer B (solid LA) was added to the reaction medium and stirring was continued for 24 h. The reaction mixture was quenched with benzoic acid (10 mg), filtered and concentrated in vacuo. Conversion was determined by 1H NMR, molar masses and dispersity of copolyesters were measured by SEC.Acknowledgements
Financial support was provided by the University of Bordeaux and the French Ministry of Research (C. T. Fellowship). The CESAMO analytic center is thanked for helpful discussions about mass spectrometry. Mrs O. Babot is gratefully acknowledged for DSC analyses.References
- F. Nederberg, E. F. Connor, M. Möller, T. Glauser and J. L.Hedrick, Angew. Chem., Int. Ed., 2001, 40, 2712–2715 CrossRef CAS.
- (a) K. E. Uhrich, S. M. Cannizzaro, R. S. Langer and K. M. Shakesheff, Chem. Rev., 1999, 99, 3181–3198 CrossRef CAS; (b) R. E. Drumright, P.R. Gruber and D. E. Henton, Adv. Mater., 2000, 12, 1841–1846 CrossRef CAS; (c) H. Tsuji, Macromol. Biosci., 2005, 5, 569–597 CrossRef CAS.
- M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093–2107 CrossRef CAS.
- (a) N. E.Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 7, 5813–5840 CrossRef; (b) D. Bourissou, S. Moebs-Sanchez and B. Martin-Vaca, C. R. Chim., 2007, 10, 775–794 CrossRef CAS.
- (a) A. F. Douglas, B. O. Patrick and P. Mahrkhodavandi, Angew. Chem., Int. Ed., 2008, 47, 2290–2293 CrossRef CAS; (b) J. Kadokawa, Y. Iwasaki and H. Tagaya, Green Chem., 2002, 4, 14–16 RSC; (c) I. Blakey, A. Yu, S. M. Howdle, A. K. Whittaker and K. J. Thurecht, Green Chem., 2011, 13, 2032–2037 RSC; (d) I. dos Santos Vieira and S. Herres-Pawlis, Eur. J. Inorg. Chem., 2012, 765–774 CrossRef CAS.
- B. G. G. Lohmeijer, R. C. Pratt, F. Leibfarth, J. W. Logan, D. A. Long, A. P. Dove, F. Nederberg, J. Choi, C. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 8574–8583 CrossRef CAS.
- C. Thomas, F. Peruch, A. Deffieux, A. Milet, J.-P. Desvergne and B. Bibal, Adv. Synth. Catal., 2011, 353, 1049–1054 CrossRef CAS.
- S. Koeller, J. Kadota, F. Peruch, A. Deffieux, N. Pinaud, I. Pianet, S. Massip, J.-M. Léger, J.-P. Desvergne and B. Bibal, Chem.–Eur. J., 2010, 16, 4197–4205 CrossRef.
- (a) M. Baidya and H. Mayr, Chem. Commun., 2008, 1792–1794 RSC; (b) N. De Rycke, F. Couty and O. R. P. David, Chem.–Eur. J., 2011, 17, 12852–12871 CrossRef CAS.
- K. Roy and P. L. A. Popelier, J. Phys. Org. Chem., 2009, 22, 186–196 CrossRef CAS.
- C. F. Lemaire, J. J. Aerts, S. Voccia, L. C. Libert, F. Mercier, D. Goblet, A. R. Pleneveaux and A. J. Luxen, Angew. Chem., Int. Ed., 2010, 49, 3161–3164 CrossRef CAS.
- O. Coulembier, P. Degée, J. L. Hedrick and P. Dubois, Prog. Polym. Sci., 2006, 31, 723–747 CrossRef CAS.
- T. Okino, Y. Hoashi and Y. Takemoto, J. Am. Chem. Soc., 2003, 125, 12672–12673 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: Mass spectra, 1H and 13C NMR, SEC and DSC analysis of representative polylactones. See DOI: 10.1039/c2ra22535b |
| This journal is © The Royal Society of Chemistry 2012 |