Interplay between hydroxyl radical attack and H-bond stability in guanine–cytosine†
José P.
Cerón-Carrasco
and
Denis
Jacquemin
*
CEISAM, UMR CNRS 6230, BP 92208, Université de Nantes, 2, Rue de la Houssinière, 44322, Nantes, Cedex 3, France. E-mail: Denis.Jacquemin@univ-nantes.fr; Fax: +33(0)2 5112 5567; Tel: +33(0)2 5112 5564
First published on 5th October 2012
Abstract
We combine ONIOM simulations with an elaborated hydrated DNA model to evaluate the effects of the hydroxyl radical, one of the major products in irradiated cells, on the stability of the guanine–cytosine (G:C) base pair. Our results demonstrate that the addition of the hydroxyl radical to the cytosine moiety results in inter-base hydrogen bonds rearrangements, eventually leading to a so-called rare tautomeric isomer. In these processes, the O6(G)–N4(C) hydrogen bond strength plays a crucial role as one of the driving forces for the double proton transfer. We also demonstrate that the surrounding environment affects the stability of the non-canonical mutagenic forms. As in undamaged DNA, the water molecules of the first hydration shell might catalyze the exchange of protons while π-stacking tunes the energetic profile of the reaction. The degenerative effects of the induced rare tautomers are shown to be governed by the transition states along the PT reactions, which in turn determines their lifetimes.
1 Introduction
The exposure of DNA to high-energy radiation induces both direct and indirect oxidative effects.1 In the former process high-energy radiation promotes the ionization of DNA in several places, e.g. sugar-phosphate backbone and base pairs.2–4 The initial damage migrates through DNA by a hopping mechanism until it is localized in one of the guanine–cytosine (G:C) base pairs, the G:C radical-trapping ability arising form guanine's low ionization potential.5–7 As shown in Fig. 1, the subsequently formed G:C•+ radical cation might react with the surrounding water molecules yielding the reduced G(8OH):C• radical.8 In the indirect mechanism the radiolysis of surrounding water molecules gives rise to an excess of highly reactive hydroxyl radicals (•OH) that tend to react with DNA in several places, including the unsaturated bonds of the G:C base pair.9–11 Although little is known about the experimental chemistry of hydroxyl radicals with DNA, recent theoretical data suggest that carbon 8 of guanine and carbon 6 of cytosine are the most favored binding sites for the formation of G:C-OH radical adducts.12 As a consequence of the apparition of such hydroxy adducts, the native double helix structure is altered leading to errors in the stored genetic information, i.e. mutations.13 Nevertheless, the •OH radical has also been associated with the biological activity of many anti-cancer agents,14 including the well-known cisplatin.15 Of course, the damaging or beneficial role played by radical adducts depends on the selectivity of the reaction: the attack of a healthy cell promotes the formation of tumors whereas drugs generate the •OH radical in cancerous cells and, consequently, are able to induce the cancer apoptosis.16 Hence the investigation of the reactions between hydroxyl radicals and DNA is crucial, not only to understand the effect of ionizing radiations, but also to explore the chemotherapeutic actions of anti-cancer drugs.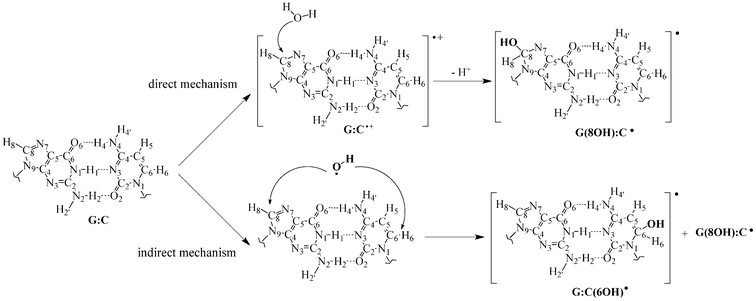 | ||
| Fig. 1 Chemical structure and atomic numbering for the canonical guanine–cytosine (G:C) base pairs as well as for the hydroxyl radical adducts formed through both direct and indirect mechanisms, G(8OH):C• and G:C(6OH)•. | ||
In that framework, considerable efforts have been devoted to disclose the possible oxidative mechanisms of DNA damage.17–20 Notably, Sevilla and co-workers have extensively studied the reactivity between radiation-produced radicals and DNA base pairs by means of both experimental and theoretical techniques.21–30 These studies have demonstrated that irradiation alters the relative stability of the canonical DNA structure by inducing proton transfer (PT) reactions in one-electron oxidized and reduced DNA bases.31–33 This conclusion is consistent with the results of Zhang and Eriksson,12 who have recently evaluated the overall reactivity between the •OH radical and one isolated G:C base pair using density functional theory (DFT). According to this seminal study, the most stable binding sites are carbon 8 of guanine and carbon 6 of cytosine, yielding the G(8OH):C• and G:C(6OH)• entities, respectively (see Fig. 1). It is noticeable that their B3LYP/6-31+G(d,p) calculations predict the presence of a rare tautomer, i.e. a structure arising from the exchange of one or more protons with respect to the original canonical form, only 4.6 kcal mol−1 above the canonical G:C(6OH)• structure.12 That picture is in sharp contrast with the non-irradiated G:C base pairs, where the tautomeric equilibrium is strongly (and hopefully) shifted in favor of the canonical form.34–42
Although there are clear hints that the number of rare tautomers could significantly increase as a consequence of the irradiation, the study of Zhang and Eriksson12 represents, to the best of our knowledge, the only theoretical approach investigating PT between the bases in G:C-OH radical adducts. Consequently, several issues remain unresolved for a full description of the interplay between irradiation and PT reactions. Specifically, since π-stacking strongly affects both the geometry and the stability of DNA base pairs,43,44 the isolated G:C-OH radical adduct model should be refined with a more realistic DNA model that accounts for stacking. In addition, explicit solvent effects need to be accurately evaluated since water molecules binding to DNA affect the hydrogen-bond of radical base pairs.45 For instance, Leszczyński and co-workers have demonstrated that the presence of water molecules might lower the energetic barrier by assisting PT process in hydrated guanine-OH adducts.46 Accordingly, explicit water molecules are mandatory to reach a balanced theoretical description of PT in DNA.
In this contribution, we use state-of-the-art ONIOM calculations to investigate the structures and relative stabilities of the two most stable DNA hydroxy adducts, G(8OH):C• and G:C(6OH)•, and the related PT reactions. To correctly appraise the influence of π-stacking in the PT, radical adducts are embedded in a hydrated doubled-stranded DNA fragment,47,48 and the solvent effects have been included through simultaneous explicit/implicit solvent models.
2 Computational details
Since all the structures studied are in radical form, the bullet superscript is omitted in the following. We have selected the same model that allowed the successful study of the rare tautomeric equilibria in undamaged DNA and its metallated forms.47–49 More precisely, the chemical environment is simulated through a double-stranded B-form trimer d(5′-GGG-3′)d(3′-CCC-5′), which includes both nucleotides moieties as well as sugar-phosphate backbone chains. The neutrality of the model is ensured by protonating the phosphate groups.47,50 The original (undamaged) DNA fragment is subsequently modified by binding the hydroxyl radical at the C8 (C6) position of guanine (cytosine), yielding to the G(8OH):C [G:C(6OH)] adduct. In that way, the G:C-OH center under study is embedded between two G:C frontier base pairs, which represents the minimal fragment able to restore π-stacking interactions in DNA.47,51 We denote this stacked DNA fragment, without explicit water molecules, as 3bp-0w. To complete the chemical model, the 3bp-0w structure is improved by including five water in the vicinity of N3(G), O6(G), N7(G), O2(C), and N4(C) atoms (see atomic numbering in Fig. 1) to explicitly mimic the first hydration shell of the G:C-OH adduct, yielding to the 3bp-5w model (see Fig. 2). This hydration shell is a suitable representation of the five solvation sites computed around GC base pairs in a fully hydrated DNA dodecamer,52,53 and agrees with the hydrogen bonds (H-bonds) pattern observed in the crystal structure.54 To complete the hydration model one can go for larger solvation shell as the one used for highly hydrated single bases.55,56 Although these studies provide important insights about single bases in solution, they can be hardly extrapolated to DNA double helix structure where both stacked bases and lateral backbone tune the hydration pattern. Furthermore, larger hydration shells would imply using less refined theoretical approach in the high-layer. Since we are mainly interested in the interbase PT processes rather than proton migration into solution, the impact of water molecules beyond the first shell is accounted for by a continuum model (see below).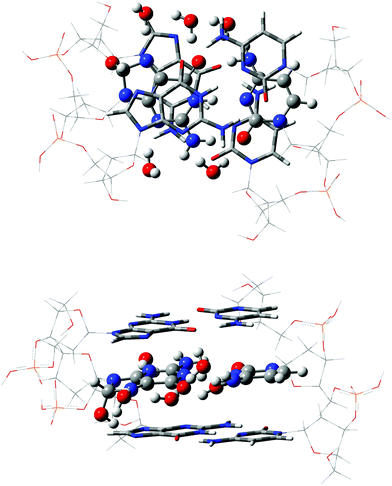 | ||
| Fig. 2 Top and side views of the 3bp-5w model for the G(8OH):C adduct. The ONIOM layers are represented with balls-and-sticks (high), tube (medium) and wireframe (low), respectively. See text for further details. | ||
Of course, the choice of the quantum mechanical model used to simulate the noncovalent interactions that dominate in DNA, namely H-bonds and π-stacking, should properly account for dispersion corrections.57–59 Within the density functional theory (DFT) approach, new functionals have been developed to assess the reactivity of (bio)chemical systems.60,61 Particularly, the hybrid meta-GGA M06-2X62,63 functional has been demonstrated to be adequate for studying these interactions in DNA.64–67 Alternatively, dispersion-corrected functionals, e.g. Grimme's B97-D,68,69 also provide highly accurate noncovalent interaction energies in a large range of problems.70–72 However, a “full” DFT simulation of the 3bp-5w model (208 atoms) is computationally too demanding, and consequently we modeled the DNA trimer by using a state-of-the-art ONIOM approach,73 which allowed us to combine different methods in the same calculation by splitting the system into up to three regions or layers: high, medium and low. The high layer contains the chemical region of the interest, namely where the reaction take places, and it is treated with the most refined theoretical level. Here, the high layer consists of the central G:C-OH radical adduct as well as the five explicit water molecules, and is described at the M06-2X/6-311G(d,p) level. The two border G:C base pairs, that confine the central G:C-OH moiety but not play an active role in the PT reactions, are placed in the medium layer and described at the B97-D/6-31G(d) level. The B97-D functional has been proved to be as efficient as M06-2X for describing the π-stacking arising from the two border base pairs while for a smaller computational cost than hybrids.74 The lateral sugar-phosphate backbones, which have a minor (direct) effect on the tautomeric equilibria,47,48 are located in the low layer and simulated with the semiempritical PM6 method.75
Since the G:C-OH adducts are doublet states, all our calculations have been performed with the unrestricted formalism for which the predicted <S2> value is a key factor. In our calculations <S2> ranges between 0.75 and 0.77 for both the high and medium layers, but can be as large as 0.84 for the low layer. However, all <S2> values become 0.75 after annihilation of the first spin contaminant, indicating that the spin contamination is not a major problem in our study. The high layer is fully optimized whereas the medium and low layers are frozen in order to ensure a physically meaningful B-DNA conformation. Analytic calculations of the vibrational frequencies confirm the nature of the optimized structures as minima (if all frequencies are real) or transition states (a single imaginary frequency). We have checked that the imaginary frequencies of the transition states (TS) involve the stretching of the transferred protons. Aiming to reach a computationally tractable model, the basis sets used during the optimizations are free of diffuse functions. Nevertheless, more accurate energies are estimated thanks to single-point ONIOM calculations performed with the M06-2X/6-31++G(d,p):B97D/6-31+G(d):PM6 approach.† These single-point calculations also include solvent effects beyond the first coordination sphere through the ONIOM-PCM/X method,76,77 which allows to combine ONIOM calculations and the integral formalism version of the polarizable continuum model (IEFPCM) by using the cavity of the whole system generated with UFF radii.78,79 Partial atomic charges were evaluated using both Mulliken and Merz-Singh-Kollman ESP schemes.80,81
3 Results and discussion
As discussed by Zhang and Eriksson,12 the G:C-OH radical adduct can exchange protons between the N1 in guanine and the N3 in cytosine, denoted N1(G) and N3(C), and between the N4 in cytosine and the O6 in guanine, N4(C) and O6(G), respectively. Therefore, to assess the effects of the OH radical on DNA stability, all possible PT paths between these atoms have been addressed. As shown in Fig. 3, the PT1 step corresponds to a single-PT reaction, where the proton H1 is transferred from N1(G) to N3(C), yielding the G(8OH):C-1 or G:C(6OH)-1 zwitterions. We emphasize that the alternative PT sequence, where the H4 proton is first transferred from N4(C) to O6(G), leads to unstable zwitterionic structures that are not stationary points. To recover the electrical neutrality, these G(8OH):C-1 or G:C(6OH)-1 zwitterions might return to the canonical structures through the reverse PT1 or, alternatively, they could undergo a double-PT reaction. As can be seen in Fig. 3 there are two possible paths for the latter mechanism: PT2 and PT3. In the PT2 path, the proton H4 is directly transferred from N4(C) to O6(G), giving the G(8OH):C-2 or the G:C(6OH)-2 structure, whereas PT3 involves a water-assisted transfer of proton H4′ leading to G(8OH):C-3 or G:C(6OH)-3. As illustrated in Fig. 4, water molecules catalyze the reaction PT3 by accepting and donating protons. We underline that in the pioneering work by Zhang and Eriksson12 the solvent effects were solely evaluated by a continuum model. The present work therefore constitutes the first study of the water-assisted tautomerism in G:C-OH adducts.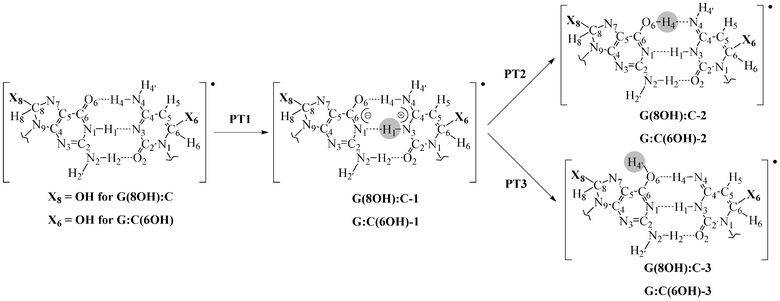 | ||
| Fig. 3 Possible paths for the inter-base proton transfer in the G(8OH):C and G:C(6OH) radical adducts. The transferred proton is circled. For the sake of clarity, only atoms of the central base pair are shown. | ||
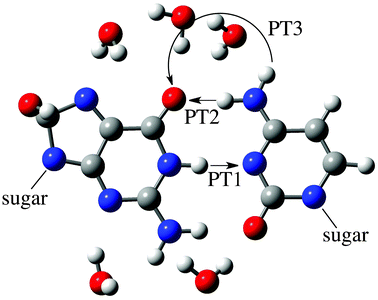 | ||
| Fig. 4 Representation of the atoms involved in the PT1, PT2 and PT3 reactions of the G(8OH):C adduct (see Fig. 3). For the sake of clarity, only the atoms in the high layer are shown, namely the G(8OH):C moiety and the water molecules of the first hydration shell. | ||
3.1 3bp-0w model
The results obtained with the 3bp-0w DNA fragment are discussed first. Since this model does not incorporate explicit water molecules, PT1 and PT2 are the only accessible processes. Both the canonical forms and its PT2 counterpart are stable minima, whereas the zwitterionic structures arising from PT1 could not be found (Table 1). Indeed, all attempts made to locate the PT1 products failed: the calculations quickly yield the G(8OH):C-2 or G:C(6OH)-2 rare tautomeric forms. The vibrational calculations reveal that G(8OH):C-2ts and G:C(6OH)-2ts structures are characterized by one imaginary frequency that can be interpreted as simultaneous PT along both N1(G)–H1–N3(C) and O6(G)–H4–N4(C) H-bonds.†Table 1 also allows to quantify the effect of the PCM corrections on the energies: they lead to a destabilization of the rare tautomers by ca. 4 kcal mol−1 for G(8OH):C-2ts and ca. 3 kcal mol−1 for G:C(6OH)-2ts with a less important impact on the rare tautomers relative energies (< 2 kcal mol−1). Despite these qualitative differences, the observed trend is not altered by the electrostatic effect of the environment. Accordingly, within the 3bp-0w model, the canonical G:C-OH adducts and the related rare tautomers are connected by one TS (with no stable intermediate), that is, the hydroxyl radical could promote a concerted double-PT between G:C base pair. Moreover, if the oxidative attack results in the formation of the G:C(6OH) adduct, a very stable rare tautomer is generated. Indeed, G:C(6OH)-2 is only 2.73 kcal mol−1 less stable than the corresponding canonical form, which clearly differs from previous calculations on undamaged DNA, where all stable rare tautomers are at least ca. 8 kcal mol−1 “above” the canonical structure.35,37,82,83 To explain such important discrepancy we have optimized the canonical structures of undamaged-DNA with the same theoretical protocol. Table 2 lists the H-bonds distances between the base pairs before and after the attack of the hydroxyl radical. Indeed, as previously demonstrated, increasing the acidity of the N1(G)–H1 bond through irradiation84,85 or metalation86,87 improves the relative stability of the rare tautomers.27,31,88,89 However, as can be deduced from Table 2, the N1(G)–N3(C) distance remains almost constant when moving from undamaged G:C to G:C-OH adducts. This hints that the acidity of the N1(G)–H1 proton is not significantly altered by the OH radical, which is in agreement with previous calculations,12 and consequently changes in the nature of this H-bond cannot account for the double-PT activation of the G:C(6OH) adduct. Indeed, the actual activation is the O6(G)–N4(C) bond. More specifically, our simulations indicate that this H-bond is shorter (0.06 Å) in the G:C(6OH) adduct than in the undamaged G:C. Although far from extreme, this change is expected to have an impact since our previous investigations demonstrated the interplay between the strength of the O6(G)–N4(C) interaction and the efficiency of the double-PT in DNA40,49,90 For instance, the elongation by 0.05 Å of the O6(G)–N4(C) bond is enough to prevent the PT2 mechanism in a single G:C base pair.40 The data collected in Tables 1 and 2 backs up our previous conclusions: (i) a strong O6(G)–N4(C) bond is a prerequisite for a effective transference of proton H1 and, consequently, for a direct double-PT reaction (PT1 + PT2) and (ii) even a slight change the strength of bond this sufficient to alter the PT mechanism.| 3bp-0w | 3bp-5w | |||
|---|---|---|---|---|
| a No stable structure in the 3bp-0w model. b Structure only accessible in the 3bp-5w model. c No stable structure neither in 3bp-0w nor in 3bp-5w. | ||||
| Structure | ΔE | ΔEPCM | ΔE | ΔEPCM |
| G(8OH):C | 0.00 | 0.00 | 0.00 | 0.00 |
| G(8OH):C-1tsa | 12.25 | 10.52 | ||
| G(8OH):C-1a | 11.97 | 7.77 | ||
| G(8OH):C-2ts | 12.84 | 17.07 | 12.64 | 10.56 |
| G(8OH):C-2 | 6.71 | 8.42 | 12.18 | 11.09 |
| G(8OH):C-3tsb | 17.98 | 18.78 | ||
| G(8OH):C-3b | 10.10 | 12.78 | ||
| G:C(6OH) | 0.00 | 0.00 | 0.00 | 0.00 |
| G:C(6OH)-1tsc | ||||
| G:C(6OH)-1c | ||||
| G:C(6OH)-2ts | 9.56 | 12.35 | 10.17 | 9.07 |
| G:C(6OH)-2 | 2.14 | 2.73 | 5.44 | 5.19 |
| G:C(6OH)-3tsb | 14.23 | 16.14 | ||
| G:C(6OH)-3b | 7.29 | 10.10 | ||
| 3bp-0w | 3bp-5w | ||||||
|---|---|---|---|---|---|---|---|
| Structure | O6(G)–N4(C) | N1(G)–N3(C) | N2(G)–O2(C) | O6(G)–N4(C) | N1(G)–N3(C) | N2(G)–O2(C) | |
| G:C (undamaged) | 2.75 | 2.96 | 2.99 | 2.79 | 2.95 | 2.96 | |
| G(8OH):C | 2.74 | 2.95 | 3.00 | 2.78 | 2.94 | 2.99 | |
| G(8OH):C-1ts | 2.55 | 2.68 | 2.97 | ||||
| G(8OH):C-1 | 2.53 | 2.81 | 3.06 | ||||
| G(8OH):C-2ts | 2.52 | 2.66 | 2.91 | 2.45 | 2.82 | 3.07 | |
| G(8OH):C-2 | 2.73 | 2.90 | 2.99 | 2.53 | 2.86 | 3.05 | |
| G(8OH):C-3ts | 2.62 | 2.87 | 3.08 | ||||
| G(8OH):C-3 | 2.92 | 2.99 | 3.01 | ||||
| G:C(6OH) | 2.66 | 2.90 | 2.99 | 2.71 | 2.91 | 2.98 | |
| G:C(6OH)-2ts | 2.49 | 2.65 | 2.92 | 2.53 | 2.66 | 2.94 | |
| G:C(6OH)-2 | 2.63 | 2.88 | 3.02 | 2.53 | 2.88 | 3.05 | |
| G:C(6OH)-3ts | 2.60 | 2.87 | 3.07 | ||||
| G:C(6OH)-3 | 2.81 | 2.95 | 3.00 | ||||
Finally, we computed the spin densities in the canonical forms by single-point full QM calculations at the M06-2X/6-311G(d,p) level. Spin localization is important because the high layer of our ONIOM model is defined as a doublet state, that is the unpaired electron lies on the hydrated G:C-OH moiety. As shown Fig. 5 the spin density is exclusively distributed in the base attacked by OH radical, that is guanine for the G(8OH):C and cytosine for G:C(6OH), which supports the reliability of the selected ONIOM partition scheme. The delocalization of the unpaired electron in DNA also confirms the mutagenic mechanism: G(8OH) and C(6OH) moieties will be the precursors of the subsequent radical reactions rather than sugar-phosphate backbones.
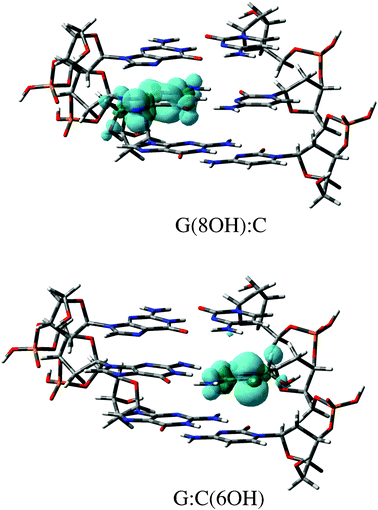 | ||
| Fig. 5 Spin density distributions (isovalue = 0.002 a.u.) of the canonical radical adducts. These M06-2X/6-311G(d,p) calculations were performed using the the 3bp-0w model. | ||
3.2 3bp-5w model
Next, the 3bp-5w model was assessed for its effect of the first hydration shell on the three PT mechanisms. First we analyze the results obtained for the G(8OH):C adduct. As seen in Table 1 the energies of both PT1 and PT2 reactions are significantly modified by the first hydration shell. Additionally, the presence of the explicit solvent molecules generates an alternative water-assisted path where water molecules (of the first hydration shell) catalyze the PT. In the ΔE profile, we have located four stationary points: the canonical G(8OH):C structure, as well as the G(8OH):C-1, G(8OH):C-2 and G(8OH):C-3 rare tautomeric forms. This contrasts with the water-free model, confirming that explicit hydration is mandatory to reach a valid description of the reactivity in DNA. The significant differences between the 3bp-0w and 3bp-5w models can be explained by the modification of the H-bonds. Indeed, the main effect of the explicit water molecules is to weaken the O6(G)–N4(C) H-bond in the canonical structure. This variation is very similar in the undamaged G:C base pair and in the G(8OH):C adduct: the O6(G)–N4(C) distance is almost the same in both structures once the hydration is accounted for. The energies listed in Table 1 also reveal that G(8OH):C-1 and G(8OH):C-2 are located in very shallow minima. The comparison of the relative energies ΔE and ΔEPCM reveals that the impact of the implicit solvent is not uniform. Indeed, the G(8OH):C-2 minimum disappears after PCM corrections (ΔEPCM), confirming its instability, whereas the G(8OH):C-1 intermediate is stabilized by ca. 3 kcal mol−1.To qualitatively evaluate the change in the G(8OH):C-2 stability, we list in Table 3 the partial atomic charges of the atoms interacting with the water molecules around the O6(G)–N4(C) H-bond. According to these charges, N7(G), O6(G) and H4′ atoms are less negative in the G(8OH):C-2 form than the canonical G(8OH):C reference. This trend is further confirmed by investigating ESP charges.† Consequently, the interaction with explicit water molecules is weaker in the G(8OH):C-2 form than in G(8OH):C which eventually results in an extra destabilization when moving from 3bp-0w to 3bp-5w. Table 1 also lists the partial atomic charges of the other two transferred protons, H4 and H1, as well as the predicted dipole moments. The charges of H4, H4′ and H1 are nearly equal in G:C and G(8OH):C. Accordingly, the hydroxyl radical does not affect the acidity of the proton in the canonical structures. Interestingly, according to Mulliken analysis the H4 proton becomes more positive in most of the non-canonical structures (with the only exception of G(8OH):C-3 and G:C(6OH)-3). In contrast, the H4′ proton carries less negative charge along with PT when the ESP model is adopted. Specifically, within the Mulliken scheme the H4 gains +0.28|e| in the G(8OH):C-2 adduct in agreement with the observed contraction (−0.25 Å) of the base pair structure during the PT2 reaction, while ESP predicts a decrease of −0.09|e|.† This finding suggests that Mulliken charges are adequate to predict qualitative changes in the base pairs geometry since they nicely correlate with the O6(G)–N4(C) bond length in the PT region.
| Structure | N7(G) | O6(G) | H4 | H4′ | H1 | dipole |
|---|---|---|---|---|---|---|
| a From PCM-(M06-2X/6-311++G(d,p):B97D/6-31+G(d):PM6) single-point calculations (see text). | ||||||
| G:C (undamaged) | −0.23 | −0.46 | 0.33 | 0.38 | 0.66 | 23.5 |
| G(8OH):C | −0.24 | −0.46 | 0.34 | 0.38 | 0.66 | 27.1 |
| G(8OH):C-1ts | −0.23 | −0.50 | 0.45 | 0.39 | 0.77 | 29.2 |
| G(8OH):C-1 | −0.23 | −0.54 | 0.49 | 0.40 | 0.67 | 29.5 |
| G(8OH):C-2ts | −0.21 | −0.49 | 0.60 | 0.38 | 0.62 | 28.0 |
| G(8OH):C-2 | −0.19 | −0.44 | 0.62 | 0.37 | 0.58 | 27.1 |
| G(8OH):C-3ts | −0.26 | −0.59 | 0.43 | 0.48 | 0.63 | 24.1 |
| G(8OH):C-3 | −0.26 | −0.54 | 0.33 | 0.34 | 0.61 | 22.7 |
| G:C(6OH) | −0.26 | −0.46 | 0.36 | 0.36 | 0.67 | 24.5 |
| G:C(6OH)-2ts | −0.27 | −0.53 | 0.49 | 0.36 | 0.79 | 26.2 |
| G:C(6OH)-2 | −0.25 | −0.49 | 0.67 | 0.34 | 0.53 | 25.0 |
| G:C(6OH)-3ts | −0.34 | −0.63 | 0.40 | 0.48 | 0.56 | 20.7 |
| G:C(6OH)-3 | −0.27 | −0.55 | 0.31 | 0.33 | 0.53 | 20.4 |
As listed in Table 3, there are also dissimilarities in the dipole moments: if the rare tautomer has a larger dipole moment than the canonical structure, the electrostatic effect of the environment stabilizes the rare tautomer and vice versa. For instance, the stabilization of the G(8OH):C-1 intermediate with the explicit–implicit solvent model originates in its enhanced polarity (dipole = 29.5 D) compared to the G(8OH):C canonical adduct (dipole = 27.1 D). In contrast, the G(8OH):C-3 (dipole = 22.7 D) is destabilized by 2.68 kcal mol−1 when adding the continuum model. This demonstrates that the description of the solvent through an explicit solvent needs to be completed with PCM corrections to reach a balanced description of the environment. Finally, the vibrational analysis allows to characterize the single imaginary frequency of the TS structures as the stretching of the N1(G)–H1–N3(C) proton for the G(8OH):C-1ts, and the stretching of the N4(C)–H4′ proton for G(8OH):C-3ts.† We therefore conclude that the formation of rare tautomers in G(8OH):C adduct proceeds through a stepwise mechanism involving first the PT1 step to yield a zwitterion and next goes through a water-assisted PT3 step that restores the neutrality.
Let us now consider the G:C(6OH) adduct. As shown in Table 1, three stationary structures have been located: the G:C(6OH) canonical form as well as the G:C(6OH)-2 and G:C(6OH)-3 rare tautomers. The interactions of the central base pair with the water molecules modify the PT reactions in the G:C(6OH) adduct: the G:C(6OH)-2 is destabilized by ca. 2.50 kcal mol−1 in the presence of the explicit water molecules. This finding is consistent with the modifications of the H-bond features when going from 3bp-0w to 3bp-5w: the water molecules weaken the O6(G)–N4(C) bond. We underline that the destabilization effect of explicit water molecules is less important in G:C(6OH)-2 than in G(8OH):C-2, which in turns hints for stronger interactions with water molecules in the former. This result also corroborates with the larger negative charges carried by the N7(G) and O6(G) in the G:C(6OH)-2 structure (see Table 3). The comparison of ΔE and ΔEPCM energetic profiles shows that the effect of the continuum solvent is related to the polarity of the rare tautomeric structure with respect to the one predicted for the canonical form: the PCM corrections stabilize G:C(6OH)-2ts and G:C(6OH)-2 and destabilize G:C(6OH)-3ts and G:C(6OH)-3. The vibrational analysis of the TS allows characterization of the tautomer-generation mechanism: the G:C(6OH)-2ts which connects G:C(6OH) to G:C(6OH)-2 presents one imaginary frequency corresponding to the stretching of the O6(G)–H4–N4(C) bond whereas, in G:C(6OH)-3ts, the normal mode associated to the imaginary frequency is the elongation of the N4(C)–H4′ bond.† Consequently, if the hydroxyl radical attacks the C6 of cytosine, the resulting G:C(6OH) adduct might lead to the formation of two rare tautomers: G:C(6OH)-2 and G:C(6OH)-3. The former is generated by a direct double-PT reaction (PT1 + PT2 steps of Fig. 3), and the latter is produced through a water-assisted double-PT (PT1 + PT3 steps of Fig. 3), both mechanisms being concerted.
3.3 Equilibrium and kinetics
As in any mutation, the macroscopic impact of the formation of rare tautomers depends on their lifetime, which are in turn mainly governed by the backward energy barrier to the canonical structure. To clarify the actual impact of the G(8OH):C-1, G(8OH):C-3, G:C(6OH)-2 and G:C(6OH)-3 rare tautomers on the stability of the genetic code, we have computed their relative free energies, equilibrium and rates constants at 298.15 K (Table 4). Specifically, the PT equilibrium constants (Keq) are calculated using:| Keq = e−ΔG0/RT | (1) |
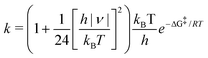 | (2) |
| Structure | ΔGb | K eq | k f | k r |
|---|---|---|---|---|
| a From PCM-(M06-2X/6-311++G(d,p):B97D/6-31+G(d):PM6) single-point calculations. b Vibrational corrections from M06-2X/6-311G(d,p):B97D/6-31G(d):PM6 optimizations. | ||||
| G(8OH):C | 0.00 | |||
| G(8OH):C-1ts | 7.52 | |||
| G(8OH):C-1 | 6.54 | 1.59 × 10−05 | 4.33 × 1007 | 2.27 × 1012 |
| G(8OH):C-3ts | 16.93 | |||
| G(8OH):C-3 | 12.59 | 5.85 × 10−10 | 3.71 × 1000 | 6.34 × 1009 |
| G:C(6OH) | 0.00 | |||
| G:C(6OH)-2ts | 5.89 | |||
| G:C(6OH)-2 | 4.33 | 1.59 × 10−05 | 7.67 × 1008 | 1.15 × 1012 |
| G:C(6OH)-3ts | 13.36 | |||
| G:C(6OH)-3 | 9.39 | 1.29 × 10−07 | 1.41 × 1003 | 1.10 × 1010 |
According to the analysis by Florián and Leszczyńki, which is based on the lifetime of a cell and the characteristic time for base-pair opening,35 a rare tautomer would be able to induce permanent genetic error(s) if the energetic barrier for forward PT is lower than 28 kcal mol−1. As shown in Table 4 all the PT reactions fulfil this first criterion. A second requisite is that the associated reverse PT barrier must be larger than 3 kcal mol−1. As expected from previous works,12 the inclusion of thermal corrections lowers the barrier heights by 1 to 3 kcal mol−1, leading to reverse ΔG‡ barriers of 0.98 kcal mol−1 [G(8OH):C-1], 4.34 kcal mol−1 [G(8OH):C-3], 1.56 kcal mol−1 [G:C(6OH)-2] and 3.97 kcal mol−1 [G:C(6OH)-3]. Consequently, while the PT3 products G(8OH):C-3 and G:C(6OH)-3 satisfy the second criterion, both G(8OH):C-1 and G:C(6OH)-2 lie below this limit and will return very quickly to the canonical forms. This is a noticeable result: the hydroxyl radical stabilizes two rare tautomers in DNA, G(8OH):C-1 and G:C(6OH)-2 (Keq∼10−05) in clear contrast to the natural tautomeric equilibrium in undamaged DNA (Keq∼10−08–10−10),34 but their degenerative effects are mitigated by their short lifetimes (kr∼1012 s−1). For the water-assisted mechanisms, the PT3 products, the G(8OH):C adduct (Keq∼10−10) is expected to promote a mutation with the same frequency as the natural spontaneous mutation in undamaged DNA, and therefore it does not alter significantly the genetic impact of PT reactions. Accordingly, the G:C(6OH)-3 rare tautomers would be the only candidates for source of a permanent mutation in G:C-OH radical adducts by PT reactions. Nevertheless, when comparing the free energies of both radical adducts in their canonical structures we observed that the G:C(6OH) form is ca. 23 kcal mol−1 less stable than the G(8OH):C form. In addition, the extremely large reverse rate constant associated with the G:C(6OH)-3 (kr∼1010 s−1) hints that this minimum would be hardly populated. Therefore our simulations show that although the OH radical attack affects the nature and relative energies of PT processes in DNA, these mechanisms cannot account significantly for the adverse effects of radicals. Since the tautomeric equilibrium of the G(8OH):C adduct is clearly shifted to its canonical structure, our calculations support the idea93 of this structure being one of the intermediates in the induced-radiation damage mechanism, more specifically a precursor of the 7,8-dihydro-8-oxoguanine, rather than primary source of mutations.
4 Conclusions and outlook
The investigation of the reaction between hydroxyl radical and DNA is crucial to understand the ionizing radiation damage as well as the biological activity of many anti-cancer drugs. In this contribution we studied the effects of the hydroxyl radical attack by exploring the stability of the two major guanine–cytosine (GC) OH radical adducts, namely G(8OH):C and G:C(6OH), and their non-canonical counterparts. Specifically, we simulated the radical-induced proton transfer (PT) reactions in both radical adducts by performing ONIOM calculations with and hydrated double-stranded DNA model. The comparison between undamaged and G:C-OH adducts demonstrated the interplay between H-bond strength and G:C-OH adducts stabilities: the attack of the hydroxyl radical on the cytosine base increases the strength of O6(G)–N4(C) bond and consequently the efficiency of the PT reactions. Our results also demonstrate that the hydroxyl induces four stable rare tautomers arising from both direct and water-assisted PT. However, these rare tautomers are expected to have a insignificant mutagenic effect due to the low back-reaction barriers to the canonical form. Of course, we cannot rule out that other cellular factors, e.g. the conformational changes imposed by the polymerases during DNA replication, affect the energetic profile of the PT reactions. Accordingly, the refinement of both environmental effects (e.g. metal ions in cells and solvent effects)55,56,94–96] and improved (larger) DNA chemical models97–100 are worthy further investigations to completely unravel the role played by PT reactions in radiation-induced DNA damage.Acknowledgements
J.P.C.C. acknowledges the fellowship provided by the Fundación Séneca, Agencia de Ciencia y Tecnología de la Región de Murcia, within its Postdoctoral Research Staff Training Program. D.J. acknowledges the European Research Council (ERC) and the Région des Pays de la Loire for financial support in the framework of Starting Grant (Marches - 278845) and a recrutement sur poste stratégique, respectively. This research used resources of the GENCI-CINES/IDRIS (Grant c2012085117), and of the CCIPL (Centre de Calcul Intensif des Pays de Loire).References
- T. Douki and J. Cadetin Radiation chemistry: from basics to applications in material and life sciences, Ed. M. Spotheim-Maurizot, M. Mostafavi, T. Douki and J. Belloni, edp sciences, 2008, pp. 177–189 Search PubMed.
- Y. A. Berlin, A. Burin and M. A. Ratner, J. Phys. Chem. A, 2000, 104, 443–445 CrossRef CAS.
- D. Khanduri, S. Collins, A. Kumar, A. Adhikary and M. D. Sevilla, J. Phys. Chem. B, 2008, 112, 2168–2178 CrossRef CAS.
- P. Schyman, L. A. Eriksson, R. bo Zhang and A. Laaksonen, Chem. Phys. Lett., 2008, 458, 186–189 CrossRef CAS.
- M. Sevilla, B. Besler and A. Colson, J. Phys. Chem., 1995, 99, 1060–1063 CrossRef CAS.
- E. Cauët, D. Dehareng and J. Liévin, J. Phys. Chem. A, 2006, 110, 9200–9211 CrossRef.
- E. Cauët and J. Liévin, J. Phys. Chem. A, 2009, 113, 9881–9890 CrossRef.
- L. I. Shukla, A. Adhikary, R. Pazdro, D. Becker and M. D. Sevilla, Nucleic Acids Res., 2004, 32, 6565–6574 CrossRef CAS.
- S. Steenken, Chem. Rev., 1989, 89, 503–520 CrossRef CAS.
- J. Cadet, T. Douki and J.-L. Ravanat, Free Radical Biol. Med., 2010, 49, 9–21 CrossRef CAS.
- J. R. Wagner and J. Cadet, Acc. Chem. Res., 2010, 43, 564–571 CrossRef CAS.
- R. B. Zhang and L. A. Eriksson, J. Phys. Chem. B, 2007, 111, 6571–6576 CrossRef CAS.
- M. S. Cooke, M. D. Evans, M. Dizdaroglu and J. Lunec, FASEB J., 2003, 17, 1195–1214 CrossRef CAS.
- S. Simizu, M. Takada, K. Umezawa and M. Imoto, J. Biol. Chem., 1998, 273, 26900–26907 CrossRef CAS.
- S. Luanpitpong, U. Nimmannit, P. Chanvorachote, S. S. Leonard, V. Pongrakhananon, L. Wang and Y. Rojanasakul, Apoptosis, 2011, 16, 769–782 CrossRef CAS.
- S. Matsugo, M. Mizuno and T. Konishi, Curr. Med. Chem., 1995, 2, 763–779 CAS.
- P. K. Shukla and P. C. Mishra, J. Phys. Chem. B, 2007, 111, 4603–4615 CrossRef CAS.
- B. H. Munk, C. J. Burrows and H. B. Schlegel, J. Am. Chem. Soc., 2008, 130, 5245–5256 CrossRef CAS.
- Q. Cheng, J. Gu, K. R. Compaan and H. F. Schaefer III, Chem.–Eur. J., 2010, 16, 11848–11858 CrossRef CAS.
- S. Kankah, J. Joseph, G. B. Schuster, R. N. Barnett, C. L. Cleveland and U. Landman, Acc. Chem. Res., 2010, 43, 280–287 CrossRef.
- M. J. E. Resendiz, V. Pottiboyina, M. D. Sevilla and M. M. Greenberg, J. Am. Chem. Soc., 2012, 134, 3917–3924 CrossRef CAS.
- V. Pottiboyina, A. Kumar and M. D. Sevilla, J. Phys. Chem. B, 2011, 115, 15090–15097 CrossRef CAS.
- A. Kumar and M. D. Sevilla, J. Phys. Chem. B, 2011, 115, 4990–5000 CrossRef CAS.
- A. Kumar, V. Pottiboyina and M. D. Sevilla, J. Phys. Chem. B, 2011, 115, 15129–15137 CrossRef CAS.
- D. Khanduri, A. Adhikary and M. D. Sevilla, J. Am. Chem. Soc., 2011, 133, 4527–4537 CrossRef CAS.
- A. Adhikary, A. Kumar, S. A. Munafo, D. Khanduri and M. D. Sevilla, Phys. Chem. Chem. Phys., 2010, 12, 5353–5368 RSC.
- A. Kumar and M. D. Sevilla, J. Phys. Chem. B, 2009, 113, 11359–11361 CrossRef CAS.
- A. Kumar, M. Sevilla and S. Suhai, J. Phys. Chem. B, 2008, 112, 5189–5198 CrossRef CAS.
- A. Kumar and M. D. Sevilla, J. Phys. Chem. B, 2006, 110, 24181–24188 CrossRef CAS.
- A. Adhikary, A. Kumar, D. Becker and M. D. Sevilla, J. Phys. Chem. B, 2006, 110, 24171–24180 CrossRef CAS.
- A. Kumar and M. D. Sevilla, Chem. Rev., 2010, 110, 7002–7023 CrossRef CAS.
- A. Adhikary, D. Khanduri and M. Sevilla, J. Am. Chem. Soc., 2009, 131, 8614–8619 CrossRef CAS.
- X. Li, Z. Cai and M. D. Sevilla, J. Phys. Chem. B, 2001, 105, 10115–10123 CrossRef CAS.
- M. D. Topal and J. R. Fresco, Nature, 1976, 263, 285–289 CrossRef CAS.
- J. Florián and J. Leszczyński, J. Am. Chem. Soc., 1996, 118, 3010–3017 CrossRef.
- J. J. Dannenberg and M. Tomasz, J. Am. Chem. Soc., 2000, 122, 2062–2068 CrossRef CAS.
- L. Gorb, Y. Podolyan, P. Dziekonski, W. A. Sokalski and J. Leszczyński, J. Am. Chem. Soc., 2004, 126, 10119–10129 CrossRef CAS.
- B. Herrera and A. Toro-Labbé, J. Phys. Chem. A, 2007, 111, 5921–5926 CrossRef CAS.
- M. Noguera, M. Sodupe and J. Bertrán, Theor. Chem. Acc., 2007, 118, 113–121 CrossRef CAS.
- J. P. Cerón-Carrasco, A. Requena, J. Zúñiga, C. Michaux, E. A. Perpète and D. Jacquemin, J. Phys. Chem. A, 2009, 113, 10549–10556 CrossRef.
- G. Villani, J. Phys. Chem. B, 2010, 114, 9653–9662 CrossRef CAS.
- G. Villani, Phys. Chem. Chem. Phys., 2010, 12, 2664–2669 RSC.
- M. Noguera, R. Ríos-Font, L. Rodríguez-Santiago, X. Solans-Monfort, A. Oliva, J. Bertran and M. Sodupe, Theor. Chem. Acc., 2009, 123, 105–111 CrossRef CAS.
- A. Gil, V. Branchadell, J. Bertran and A. Oliva, J. Phys. Chem. B, 2009, 113, 4907–4914 CrossRef CAS.
- H. M. Jaeger and H. F. Schaffer-III, J. Phys. Chem. B, 2009, 113, 8142–8148 CrossRef CAS.
- J. Gu and J. Leszczyński, J. Phys. Chem. A, 1999, 103, 577–584 CrossRef CAS.
- H.-Y. Chen, C.-L. Kao and S. C. N. Hsu, J. Am. Chem. Soc., 2009, 131, 15930–15938 CrossRef CAS.
- H.-Y. Chen, S.-W. Yeh, S. C. N. Hsu, C.-L. Kao and T.-Y. Dong, Phys. Chem. Chem. Phys., 2011, 13, 2674–2681 RSC.
- J. P. Cerón-Carrasco, J. Zúñiga, A. Requena, E. A. Perpète, C. Michaux and D. Jacquemin, Phys. Chem. Chem. Phys., 2011, 13, 14584–14589 RSC.
- D. M. Close and K. T. Øhman, J. Phys. Chem. A, 2008, 112, 11207–11212 CrossRef CAS.
- A. Galano and J. R. Alvarez-Idaboy, Phys. Chem. Chem. Phys., 2012, 14, 12476–12484 RSC.
- P. Auffinger and E. Westhof, J. Mol. Biol., 2000, 300, 1113–1131 CrossRef CAS.
- V. Makarov, B. Pettitt and M. Feig, Acc. Chem. Res., 2002, 35, 376–384 CrossRef CAS.
- B. Schneider and H. M. Berman, Biophys. J., 1995, 69, 2661–2669 CrossRef CAS.
- C. Alemán, Chem. Phys., 1999, 244, 151–162 CrossRef.
- C. Alemán, Chem. Phys., 2000, 253, 13–19 CrossRef.
- K. S. Thanthiriwatte, E. G. Hohenstein, L. A. Burns and C. D. Sherrill, J. Chem. Theory Comput., 2011, 7, 88–96 CrossRef CAS.
- C. Dupont, C. Patel and E. Dumont, J. Phys. Chem. B, 2011, 115, 15138–15144 CrossRef CAS.
- S. N. Steinmann and C. Corminboeuf, J. Chem. Theory Comput., 2011, 7, 3567–3577 CrossRef CAS.
- L. Goerigk and S. Grimme, J. Chem. Theory Comput., 2010, 6, 107–126 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, J. Chem. Theory Comput., 2011, 7, 669–676 CrossRef CAS.
- Y. Zhao, N. E. Schultz and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- J. Gu, J. Wang, J. Leszczynski, Y. Xie and H. F. Schaefer III, Chem. Phys. Lett., 2008, 459, 164–166 CrossRef CAS.
- J. C. Hargis, H. F. Schaefer III, K. N. Houk and S. E. Wheeler, J. Phys. Chem. A, 2010, 114, 2038–2044 CrossRef CAS.
- C. Acosta-Silva, V. Branchadell, J. Bertran and A. Oliva, J. Phys. Chem. B, 2010, 114, 10217–10227 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Chem. Phys. Lett., 2011, 502, 1–13 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1997, 107, 8554–8560 CrossRef CAS.
- J. Antony and S. Grimme, Phys. Chem. Chem. Phys., 2006, 8, 5287–5293 RSC.
- J. Wu, L. Albrecht and R. J. Boyd, J. Phys. Chem. B, 2011, 115, 14885–1890 CrossRef CAS.
- J. Wu, V. E. J. Walker and R. J. Boyd, J. Phys. Chem. B, 2011, 115, 3136–3145 CrossRef CAS.
- S. Dapprich, I. Komáromi, K. S. Byun, K. Morokuma and M. J. Frisch, THEOCHEM, 1999, 461–462, 1–21 CrossRef CAS.
- J. P. Cerón-Carrasco, A. Requena and D. Jacquemin, Theor. Chem. Acc., 2012, 131, 1188 CrossRef.
- J. J. P. Stewart, J. Mol. Model., 2007, 13, 1173–1213 CrossRef CAS.
- T. Vreven, B. Mennucci, C. O. da Silva, K. Morokuma and J. Tomasi, J. Chem. Phys., 2001, 115, 62–72 CrossRef CAS.
- S. J. Mo, M. Vreven, B. Mennucci, K. Morokuma and J. Tomasi, Theor. Chem. Acc., 2004, 111, 154–161 CrossRef CAS.
- J. Tomasi, M. Mennucci and E. Cances, J. Mol. Struct. (Theochem), 1999, 464, 211–226 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS.
- U. C. Singh and P. A. Kollman, J. Comput. Chem., 1984, 5, 129–145 CrossRef CAS.
- B. H. Besler, K. M. Merz and P. A. Kollman, J. Comput. Chem., 1990, 11, 431–439 CrossRef CAS.
- A. Abo-Riziq, L. Grace, E. Nir, M. Kabelac, P. Hobza and M. S. de Vires, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 20–23 CrossRef CAS.
- G. Villani, Chem. Phys., 2006, 324, 438–446 CrossRef CAS.
- K. Kobayashi and S. Tagawa, J. Am. Chem. Soc., 2003, 125, 10213–10218 CrossRef CAS.
- K. Kobayashi, R. Yamagami and S. Tagawa, J. Phys. Chem. B, 2008, 112, 10752–10757 CrossRef CAS.
- B. Song, J. Zhao, R. Griesser, C. Meiser, H. Sigel and B. Lippert, Chem.–Eur. J., 1999, 5, 2374–2387 CrossRef CAS.
- G. Schröder, B. Lippert, M. Sabat, C. J. L. Lock, R. Faggiani, B. Song and H. Sigel, J. Chem. Soc., Dalton Trans., 1995, 3767–3775 RSC.
- J. P. Cerón-Carrasco, A. Requena, C. Michaux, E. A. Perpète and D. Jacquemin, J. Phys. Chem. B, 2010, 114, 13439–13445 CrossRef.
- J. P. Cerón-Carrasco and D. Jacquemin, ChemPhysChem, 2011, 12, 2615–2623 CrossRef.
- J. P. Cerón-Carrasco, A. Requena, C. Michaux, E. A. Perpète and D. Jacquemin, J. Phys. Chem. A, 2009, 113, 7892–7898 CrossRef.
- E. Z. Wigner, Phys. Chem., 1941, 19, 203–216 Search PubMed.
- G. A. DiLabio and E. R. Johnson, J. Am. Chem. Soc., 2007, 129, 6199–6203 CrossRef CAS.
- J. Reynisson and S. Steenken, Phys. Chem. Chem. Phys., 2002, 4, 527–532 RSC.
- N. Russo, M. Toscano and A. Grand, J. Am. Chem. Soc., 2001, 123, 10272–10279 CrossRef CAS.
- N. Russo, M. Toscano and A. Grand, J. Phys. Chem. B, 2001, 105, 4735–4741 CrossRef CAS.
- N. Russo, M. Toscano and A. Grand, J. Phys. Chem. A, 2003, 107, 11533–11538 CrossRef CAS.
- P. F. Loos, E. Dumont, A. Laurent and X. Assfeld, Chem. Phys. Lett., 2009, 475, 120–123 CrossRef CAS.
- D. Ambrosek, P. F. Loos, X. Assfeld and C. Daniel, J. Inorg. Biochem., 2010, 104, 893–901 CrossRef CAS.
- E. Cauët, M. Valiev and J. H.Weare, J. Phys. Chem. B, 2010, 114, 5886–5894 CrossRef.
- J. Garrec, C. Patel, U. Rothlisberger and E. Dumont, J. Am. Chem. Soc., 2012, 134, 2111–2119 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: (1) Comparison of ONIOM energies computed at M06-2X/6-31G(d,p):B97D/6-31G(d):PM6 and M06-2X/6-31++G(d,p):B97D/6-31+G(d):PM6 levels; (2) Imaginary vibrational frequencies and normal modes analysis for transitions states; (3) Summary of ESP charges. See DOI: 10.1039/c2ra22389a |
| This journal is © The Royal Society of Chemistry 2012 |