Oligonucleotide solid-phase synthesis on fluorescent nanoparticles grafted on controlled pore glass†
Gabriel De
Crozals
a,
Carole
Farre
a,
Grégoire
Hantier
a,
Didier
Léonard
a,
Christophe A.
Marquette
b,
Céline A.
Mandon
b,
Laurence
Marmuse
c,
Cédric
Louis
c,
Jean-Jacques
Toulmé
d,
Claire
Billotey
e,
Marc
Janier
e and
Carole
Chaix
*a
aUniversité de Lyon, UMR CNRS 5280, ISA, France. E-mail: carole.chaix-bauvais@univ-lyon1.fr
bUniversité de Lyon, UMR CNRS 5246, ICBMS, France
cNANO-H S.A.S., Lyon, France
dUMS3033-US001, IECB, Bordeaux, France
eUniversité de Lyon, UMR CNRS 5620, LPCML, Médecine nucléaire, Hôpital E. Herriot, France
First published on 5th October 2012
Abstract
Oligonucleotide solid-phase synthesis is now possible on nano-sized particles, thanks to the use of controlled pore glass-nanoparticle assemblies. We succeeded in anchoring silica nanoparticles (NPs) inside the pores of micrometric glass via a reversible covalent binding. The pore diameter must be at least six times the diameter of the nanoparticle in order to maintain efficient synthesis of oligonucleotides in the synthesizer. We demonstrated that the pores protect NP anchoring during DNA synthesis without decreasing the coupling rate of the phosphoramidite synthons. This bottom-up strategy for NP functionalization with DNA results in unprecedented DNA loading efficiency. We also confirmed that the DNA synthesized on the nanoparticle surface was accessible for hybridization with its complementary DNA strand.
Introduction
Nanomaterials have emerged as a research field of great interest for biomedical applications. In medicine, the scope of applications ranges from in vivo imaging, diagnosis to therapeutics.1–3 Furthermore, nanometer-sized materials have proved very useful for biosensing and in vitro labelling thanks to their high surface coverage that significantly improves detection sensitivity.4,5For all these applications, silica has been intensively studied and has proved to be a material of choice owing to its biocompatibility, nonimmunogenicity, easy functionalization and chemical, thermal, and mechanical stability.6,7
While nanoparticles (NPs) have their own intrinsic properties, for biological applications it is generally necessary to impart additional properties or functions through physical or chemical coupling between a NP and one or more molecules. This is often true in the case of synthetic materials, where the NP enables detection but requires conjugation to a biological moiety for targeting or bioactivity.8,9 The great potential of functionalized nanoparticles prompted us to think about how to optimize the surface functionalization of an inorganic nano-sized particle with a complex biomolecule. Indeed, the formation of these conjugates is usually restricted by the coupling step because of problems related to non-specific adsorption, aggregation, purification and low yield.10
In this context, we focused on developing a new strategy to obtain oligonucleotide functionalized particles that could be used to target DNA or protein. We considered the possibility of performing direct and automated oligonucleotide solid-phase synthesis on nano-sized silica supports. Silica is the standard material for this purpose because it can be easily derivatized with the functional arm that serves to initiate solid-phase synthesis. However, because of technical considerations associated with automated DNA synthesis (notably the high pressure in the instrument and the porosity of the filters closing the synthesis column), the solid supports must be micrometer sized to be held in the synthesizer column during DNA synthesis.
In a previous paper, we developed an innovative silica material called NOM (nano-on-micro): a support that allows direct oligonucleotide synthesis on nano-sized particles immobilized on a micrometric support.11 Seliger et al. also developed a similar “support on support” concept, based on the reversible anchoring of NPs to the surface of polymeric micrometric beads.12 The common point of the two strategies was that the core particle was of a micrometric size, non-porous and inert under the conditions of oligonucleotide synthesis. However, the main drawback of these approaches was the loss of NPs anchored on the micrometric support during DNA synthesis because of mechanical friction occurring inside the synthesis column. It resulted in very low yields of DNA-NPs.
Herein, we report the use of controlled pore glass (CPG) as a micrometric support for holding NPs. The advantages of CPG are its homogeneous pore size and high specific surface, allowing efficient coverage with NPs outside and inside the pores. CPG is widely available with different pore sizes and surface areas. In our strategy, the pore size must be adapted to the size of the NPs to be held. We estimated that the pore diameter must be at least six times the diameter of the NPs in order to maintain efficient synthesis of oligonucleotides in the synthesizer. We describe DNA solid-phase synthesis on 50 nm NPs supported on CPG with a pore diameter of 300 nm. Rhodamine encapsulated in the core of the nanoparticles during their fabrication leads to a highly fluorescent silica nanomaterial. The reversible grafting of NPs on a CPG surface was achieved via ester bonding. At the end of DNA synthesis, mild basic conditions were applied to release the NPs. The integrity of the oligonucleotide-functionalized NPs after release was confirmed by Transmission Electron Microscopy (TEM), Surface Plasmon Resonance imaging (SPRi) and Atomic Force Microscopy (AFM). Our strategy provided fluorescent NPs highly functionalized with synthetic oligonucleotides. High loaded DNA-NPs could have very interesting in vivo properties, especially nuclease resistance and increased cellular uptake.13,14
Results and discussion
NP grafting on controlled pore glass
It is well-established that CPG is a suitable support for automated DNA synthesis.15 This material is chemically inert and behaves well in the presence of solvents and reagents during synthesis. Its intrinsic porosity offers an extremely high surface area which allows high loading of functional groups or biomolecules. In this work, we aim to load silica nanoparticles on the CPG surface in order to achieve automated oligonucleotide synthesis on the NP surface.Automated oligonucleotide synthesis relies on the use of solid supports entrapped in synthesis columns closed by filters, and to date, there are no commercially available filters displaying such low porosity (lower than 50 nm) and able to resist the reagents and pressure used in a DNA synthesizer. To grow oligonucleotides on nano-sized materials, they must be anchored on a micrometric support retained within the columns of the DNA synthesizer. We have developed a reversible anchoring strategy to maintain the silica NPs grafted on CPG during automated DNA synthesis and to release DNA-NP conjugates by means of specific treatment at the end of synthesis (Fig. 1).
 | ||
| Fig. 1 The different steps of the strategy developed for oligonucleotide synthesis on nanoparticles and electron microscopy photos of the corresponding materials. (A) TEM image of NPs before grafting on CPG. (B) SEM image of NP-CPG after DNA synthesis. (C) TEM image of DNA-NP conjugates after release from the support. | ||
First, fluorescent silica NPs were silanized with the 3-(triethoxysilyl)propyl-hydroxyhexyl urea (TESPHU), a hydroxysilane suitable for DNA synthesis. The TEM image in Fig. 1A shows well-dispersed silanized nanoparticles. In parallel, the CPG support was derivatized with 3-aminopropyltriethoxysilane (APTES) under acidic conditions. In a second step, amines were converted to COOH functions by a succinic anhydride reaction to allow subsequent ester bond formation with hydroxyl-functionalized nanoparticles. Then, a capping step was carried out with piperidine to block the residual activated ester functions from CPG. SEM analysis of the resulting assemblies revealed that NPs had been efficiently grafted inside the pores of CPG beads (Fig. S1 in the ESI†).
The NPs initially grafted on the external surface of the CPG had disappeared, because of the mechanical friction of the material during the capping step. The pores of CPG have the great advantage of protecting the NP from mechanical release, which often occurs during reaction stirring. Optical microscopy observations showed the homogeneous functionalization of CPG beads with fluorescent NPs (Fig. S2 in the ESI†).
The loading yield on CPG was estimated by quantifying the NPs remaining in the solution after the grafting reaction. The absorbance of non-reacting NPs was recorded by a UV-visible spectrophotometer at 554 nm (maximum rhodamine absorption). This value was compared to a standard calibration curve obtained with different dilutions of a reference NP solution of 9.3 × 1013 ± 9 × 1012 NP mL−1 (Fig. S3 in the ESI†). Thus, the percentage of grafted NPs was estimated at 43 ± 9% of the initial solution, which represents 3.2 × 1014 ± 7 × 1013 NP per gram of CPG.
DNA synthesis
DNA synthesis was initiated from these NP-CPG assemblies. Hydroxyl groups from the NPs were able to initiate oligonucleotide synthesis. DNA solid-phase synthesis was achieved via standard phosphoramidite chemistry, as described by Beaucage et al. in 1992.16Thanks to the large volume of the pores (300 nm diameter), a 23mer HBV (hepatitis B virus) DNA probe was synthesized on the assemblies using a standard 1 μmol scale coupling program with yields comparable to conventional support.
Information can be extracted from the experimental data, in particular from coupling yields. Measurement was performed by quantifying the dimethoxytrityl (DMT) group of the phosphoramidite synthon, after its coupling to the NP-CPG support. The synthesis was highly reproducible. Coupling of the first phosphoramidite synthon on five different NP-CPG batches provided similar loading of 7.8 μmol g−1 with a low standard deviation. Then, DNA synthesis was carried out and coupling yields were calculated at different steps. The values were corrected by withdrawing the background noise resulting from slight nonspecific coupling of the phosphoramidite synthon on CPG (less than 10%). These data are reported in Table 1.
| Coupling step | X (μmol g−1)a | OY (%)b | AY (%)c |
|---|---|---|---|
| a X = Support loading at the different steps of NP functionalization by PEG and DNA. b OY = overall coupling yield. c AY = average coupling yield per incorporation (average value and standard deviation of five independent syntheses). | |||
| 1st PEG | 7.8 ± 0.4 | 100 | N/A |
| 5th PEG | 5.4 ± 0.2 | 69 ± 3 | 92.8 |
| DNA | 3.8 ± 0.4 | 48 ± 4 | 98.5 |
Overall yield (OY) represents the proportion of the growing chains, and is calculated as follows:
| OY = 100 × (Xn/X1) |
Where Xn is the support loading at the n incorporation, X1 is support loading at the 1st incorporation.
We found that PEG spacer incorporation before DNA synthesis improved the quality of the DNA probe. The number of growing chains decreased by 31% during the first five PEG incorporations, which is equivalent to an average coupling yield per incorporation (AY) of 92.8%. AY from the fifth to the last synthon incorporation, which corresponds to the 23mer DNA synthesis, was 98.5%. It should be noted that the 48% OY refers to the entire synthesis and corresponds to the incorporation of the 5 PEG and all phosphoramidite synthons of the 23mer DNA. Consequently, the overall yield of the DNA synthesis is estimated at 70% after withdrawal of the loss of yield resulting from PEG incorporations on NP-CPG.
Microscopic observations were performed to control the stability of the assemblies. A SEM photo of the NP-CPG assemblies after DNA synthesis is shown in Fig. 1B. It is worth noting that the cavity of the pores protected the NPs throughout the DNA synthesis. This support ensures the colloidal stability of the NPs during DNA synthesis by protecting them from mechanical friction resulting from the flow of reagents and argon in the synthesizer column.
After DNA synthesis, the nucleic bases were deprotected with potassium carbonate 0.05 M in methanol and NPs were released from the CPG by an aqueous ammonia treatment. A complete study of DNA-NP stability in ammonia was achieved by TEM to determine the basic conditions allowing NP release from CPG without any degradation of the particles. It appeared that NPs were stable in 1% NH4OH at 60 °C over a period of 2 h (Fig. S4 in the ESI†) after which, a slow degradation of the silica material was observed by TEM. The release protocol was determined on the basis of this study. A solution of 1% NH4OH in water was added to the assembly. The reaction was carried out for 20 min at 60 °C under gentle stirring and then immediately neutralized with an acetic acid (0.5 M) solution to reach pH 7, when the reaction was stopped. This release step was repeated four or five times until the release of NPs from the CPG was complete. We assume that this mild basic treatment prevents any degradation of silica NPs and allows the perfect integrity of the oligonucleotide-functionalized NPs (Fig. 1C).
Top surface analysis of nanoparticles was performed using Time-of-Flight Secondary Ion Mass Spectrometry (ToF-SIMS) in positive and negative mode on silica NP, TESPHU-NP and DNA-NP samples (Table 2).
| m/z | Detected ion | Sample | |||
|---|---|---|---|---|---|
| Exp. | Calc. | A | B | C | |
| 27.97 | 27.98 | Si+ | X | X | |
| 28.03 | 28.03 | C2H4+/CH2N+ | X | ||
| 30.03 | 30.03 | CH4N+ | X | X | |
| 44.97 | 44.98 | SiOH+ | X | X | |
| 45.04 | 45.03 | C2H5O+ | X | ||
| 112.07 | 112.05 | C4H6N3O+ (cytosine) | X | ||
| 118.11 | 118.12 | C6H16NO+ (TESPHU arm) | X | ||
| 136.08 | 136.06 | C5H6N5+ (adenine) | X | ||
| 152.09 | 152.06 | C5H6N5O+ (guanine) | X | ||
| 26.00 | 26.00 | CN− | X | X | |
| 42.00 | 42.00 | CNO− | X | X | |
| 62.97 | 62.96 | PO2− | X | ||
| 78.97 | 78.96 | PO3− | X | ||
For silica NPs, only peaks characteristic of silica were detected. After NP modification with TESPHU (sample B), silica was still detected together with a peak characteristic of the TESPHU derivative (C6H16NO+, detected at m/z: 118.11). Molecular peaks containing nitrogen and carbon atoms were identified in both positive and negative ion modes. Interestingly, in the DNA-NP sample C, peaks related to silica and to TESPHU disappeared, masked by the additional layer of DNA. The ToF-SIMS information depth being limited to the top first monolayer(s), it indicates a significant grafting at the top surface of nanoparticles. For instance, the SiOH+ ion (at m/z: 44.97) is poorly detected while C2H5O+ (at m/z: 45.04) related to the sugar moiety is strongly detected. In the 110–160 m/z range, characteristic peaks of DNA were also detected and identified as cytosine, adenine and guanine specific signatures as previously indicated in the literature.17 Thymine was not detected due to its low ionization probability. In addition, phosphate related peaks were identified in the negative mode (See Fig. S5 in the ESI†). ToF-SIMS allowed us to follow the different steps of NP functionalization thanks to its high surface sensitivity and spectral fingerprinting (characteristic molecular peaks).
To be able to analyse the quality of the oligonucleotide synthesized from the NP-CPG assemblies, we incorporated a base-cleavable sulfone linker at the first step of the oligonucleotide synthesis. Consequently, the basic K2CO3 treatment achieved at the end of the process for deprotection also released the oligonucleotides in solution by cutting the sulfone link. The crude DNA material was analysed by HPLC. The chromatogram in Fig. 2 confirms the quality of the oligonucleotide synthesized from NP-CPG assemblies. 23mer purity was estimated at 68% by integrating the peak area of the full length oligonucleotide on the chromatogram. The expected structure of HBV DNA 3′-(PEG)1-phosphate was confirmed by MALDI-ToF mass spectrometry (Fig. S6 in the ESI†).
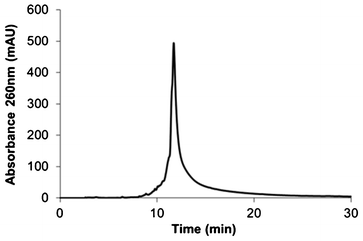 | ||
| Fig. 2 HPLC chromatogram of the crude HBV DNA synthesized from NP-CPG. | ||
DNA surface coverage
To study the surface functionalization, the gain of size resulting from the oligonucleotide layer was estimated by Dynamic Light Scattering (DLS) measurements. The length of a 23-base oligonucleotide under a coiled rod-like structure was estimated at 6.9 nm (0.3 nm per base, as cited by Mirkin et al.).18The lengths of TESPHU and single PEG spacers were estimated at 2.6 nm and 1.2 nm respectively (Fig. 3). The resulting theoretical value of the overall DNA-functionalized layer was 10.7 nm. To validate this assumption experimentally, the size and distribution of NPs were measured by Dynamic Light Scattering (Fig. S7 in the ESI†). The hydrodynamic diameters of NP and NP-TESPHU-(PEG)1-oligonucleotide in water were 67 nm and 82 nm respectively, with excellent polydispersity indexes (PdI) of 0.064 and 0.088. It is worth noting that these diameters obtained through DLS measurements with fluorescent NPs cannot be interpreted as correct average hydrodynamic diameters since this technique is not suitable for fluorescent samples. However, hydrodynamic diameter variations can be compared. The diameter gain measured by DLS indicated a TESPHU-(PEG)1-oligonucleotide layer of 7.5 nm that is not far from the theoretical value of 10.7 nm. We assume that approximately half of the NP surface was properly covered by full length oligonucleotides, since the NP portion facing CPG did not initiate oligonucleotide synthesis. The overall hydrodynamic diameter measured by DLS should be smaller on average than that of a fully covered NP with a TESPHU-(PEG)1-oligonucleotide layer. In conclusion, DLS measurements corroborate, with reasonable accuracy, the calculated value of the NP-TESPHU-(PEG)1-oligonucleotide diameter.
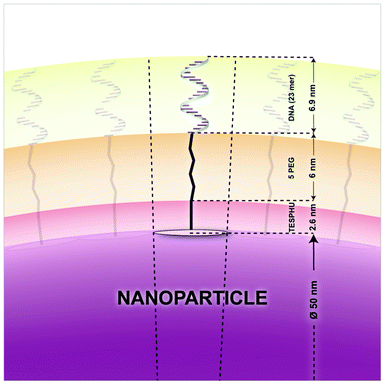 | ||
| Fig. 3 Schematic representation of the nanoparticle surface covered by DNA. The circle on the NP surface represents the DNA footprint. | ||
Another important subject of this work is the amount of DNA synthesized on the surface of each NP. In order to calculate the surface coverage of DNA strands, we made certain assumptions. Firstly, nanoparticles were simplified to perfect spheres of 50 nm. Secondly, we estimated the proportion of the nanosphere surface covered by full-length oligonucleotides. To do so, the surface of the nanoparticle was divided in three areas: 1) non-functionalizable, 2) functionalized with low yields (first incorporated phosphoramidites), 3) functionalized with good yields (complete DNA strands). From SEM observations on the packing of NPs on CPG, we assessed the non-functionalizable part of each NP to be 10% of the total surface. This portion represents the non-reactive area where hydroxyl arms are either connected with the support or not accessible due to steric hindrance. The percentage of the second area with low access, partially functionalized, was calculated from trityl assays (Table 1). At the fifth incorporation, we observed a clear inflection of phosphoramidite coupling yields. Up to this inflection point, AY was 92.8%. At this point, the average coupling yield per incorporation started to increase to reach 98.5%. We hypothesized that phosphoramidites could no longer access the second area facing CPG because of steric hindrance induced by the first five incorporations and DNA synthesis started with maximum yield on the perfectly accessible surface of the NPs. Before this inflection point, the OY decreased by 30% (incorporation of the 5 PEG with low AY). Maximum AY was measured when DNA synthesis started. Considering these assumptions, we estimated that full length DNA covered an area representing 60% of the total surface area of the NPs.
Considering NP loading on the support and DNA loading on the NPs as calculated above, the surface coverage of DNA on NPs was 248 ± 25 pmol cm−2 (on 60% of the NP surface), which represents 1.50 ± 0.15 oligo nm−2 (on 60% of the NP surface). An average number of 7050 DNA strands per nanoparticle was calculated. To the best of our knowledge, such high functionalization has never been reported in the literature (Table 3).
| NP material | DNA | DNA per NPa | Maximum surface coverageb | Reference | |
|---|---|---|---|---|---|
| DNA nm−2 | pmol cm−2 | ||||
| a Number of DNA strands per NP calculated for a 50 nm diameter NP. b Surface coverage usually expressed either in number of DNA strands per square nanometer or picomoles per square centimeter. c On 60% of the NP surface. | |||||
| silica | 23mer-(PEG)5 | 7050 | 1.50c | 248c | this work |
| silica | 42mer-NH2 | 126 | 0.016 | 2.7 | Wang et al.23 |
| silica | 24mer-SH | 322 | 0.041 | 6.8 | Wu et al.24 |
| silica | 12mer-NH2 | 589 | 0.075 | 12.5 | Rao et al.25 |
| gold | 37mer-SH | 1100 | 0.14 | 23.2 | Zhao et al.26 |
| gold | 28mer-SH | 1414 | 0.18 | 29.9 | Jones et al.27 |
| gold | 25mer-SH | 640 | 0.081 | 13.5 | Hill et al.18 |
| gold | 15mer-PEG-SH | 1257 | 0.16 | 26 | Hurst et al.22 |
| silver | 12mer-SH | 6832 | 0.87 | 144.7 | Dougan et al.28 |
The functionalization of NPs with hydroxysilanes like TESPHU is not common in the literature because of the instability of these compounds. The coupling of the first phosphoramidite actually gave us some data about TESPHU silanization.
With a loading of 7.8 μmol g−1 for the first PEG, TESPHU surface coverage was about 2.1 OH functions per nm2 (considering 90% of the NP surface). Studies of APTES silanization are more widespread in the literature. For instance, Giaume et al. studied the coverage of amine functions and their availability for protein coupling.19 They reported a high surface loading of 28–51 NH2/nm2 but only 1.9 amino groups per nm2 were accessible for the fluorescein coupling reaction. This is in good agreement with our functionalization measurements and those reported by other groups.20,21
To evaluate the crowding of DNA strands (on 60% of the NP surface), it is convenient to use the term of “footprint”, first introduced by Mirkin et al.18 The footprint represents the average area each oligonucleotide occupies on the nanoparticle surface (Fig. 3). By dividing the surface area covered by DNA by the number of strands, we found a footprint of 0.67 nm2. This footprint value is 20 times less than that reported by the Mirkin group for a 50 nm gold particle (12 nm2).18
DNA loading on NPs is restricted by steric hindrance, possible electrostatic repulsion and nonspecific adsorption of DNA bases, however, our bottom-up strategy dramatically reduces these limitations and provides more functionalized DNA-NPs than top-down strategies.22 The major advantage lies in the step-by-step construction of the DNA structure. It appears that it is much easier to couple a small molecule, like a phosphoramidite building block, than an entire biomolecule, like DNA. The lower coupling yield of the first incorporations shows that steric effects are not totally suppressed, however phosphoramidite incorporation remains efficient (>90%). This method also avoids electrostatic repulsion by using cyanoethyl-protected phosphates instead of negatively charged phosphates.
Considering the high density of DNA that results from our strategy, it is necessary to raise the issue of the accessibility of these DNA strands for specific binding with the complementary targets. We assume that synthetic DNA strands have a linear extended conformation and almost perpendicular orientation at the surface in an aqueous buffer since they are closely packed on the surface.29 A hybridization experiment was performed to study DNA accessibility.
DNA hybridization assay
A hybridization test was performed with the complementary target and monitored by Surface Plasmon Resonance imaging (SPRi). The hybridization reaction of DNA-NPs with their complementary strands occurred a few seconds after injection. No signal variation was detected on the area functionalized with non-complementary strands. A second injection of a more concentrated solution of DNA-NP led to saturation of the surface. This clearly proved the success of DNA synthesis and secondly, the accessibility of the synthetic DNA present on the NP surface. Moreover, Atomic Force Microscopy (AFM) and fluorescence microscopy showed that the immobilization of the DNA-NP conjugates only occurred on the area spotted with the complementary DNA strands (Fig. 4) as no particles and no fluorescence were observed either on the non-complementary spot (polyT 23mer) or on the non-functionalized gold surface. The size of the NPs observed by AFM on the functionalized positive spot corresponded to single particles grafted on the surface. No aggregates were observed by AFM and fluorescence microscopy.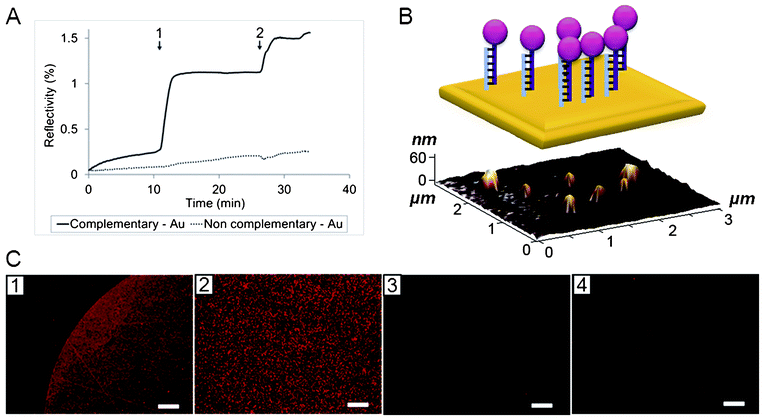 | ||
| Fig. 4 (A) SPRi of hybridization assay (1: injection of DNA-NP solution, dilution 1/100, 2: injection of DNA-NP solution, dilution 1/25). (B) 3 × 3 μm AFM image of DNA-NP grafted on the complementary spot (bottom) and its schematic representation (top). (C) Fluorescence microscopy of SPR prism (1 and 2: border and middle of the complementary spot, 3: non-complementary spot, 4: gold); scale bars are 100 μm for images 1, 3 and 4 and 20 μm for image 2. | ||
Experimental section
Materials
6-Aminohexan-1-ol, 3-(triethoxysilyl)propyl isocyanate, 3-aminopropyl-triethoxysilane (APTES), 28% ammonium hydroxide in water (NH4OH), triethylamine, dimethylaminopyridine (DMAP), succinic anhydride, 4-(carboxymethyl)aniline (CMA), phosphate buffer saline (PBS), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS), sodium nitrite, bovine serum albumin (BSA), Tween 20 and O-benzotriazol-1-yl-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU) were purchased from Sigma-Aldrich (Saint Quentin Fallavier, France). Isopropanol, dimethylformamide (DMF) and 1 M hydrochloric acid were purchased from SDS (Peypin, France). 68% nitric acid was purchased from VWR (Strasbourg, France).The following ultramild deprotection nucleoside CE-phosphoramidite synthons were used for DNA synthesis: 5′-dimethoxytrityl-N-phenoxyacetyl-2′-deoxyadenosine, 3′-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphoramidite (Pac-dA-CE phosphoramidite), 5′-dimethoxytrityl-N-acetyl-2′-deoxycytidine, 3′-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphoramidite (Ac-dC-CE phosphoramidite), 5′-dimethoxytrityl-N-isopropyl-phenoxy- acetyl-deoxyguanosine, 3′-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphoramidite (iPr-Pac-dG-CE phosphoramidite), and 5′-dimethoxytrityl-2′-deoxythymidine, 3′-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphoramidite (dT-CE phosphoramidite), 2-[2-(4,4′-dimethoxytrityloxy)ethylsulfonyl]ethyl-(2-cyanoethyl)-(N,N-diisopropyl)-phosphoramidite (chemical phosphorylation reagent), 9-O-dimethoxytrityl-triethyleneglycol 1-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphoramidite (PEG) and all the oligonucleotide synthesis reagents (i.e., activator solution (0.45 M tetrazole in acetonitrile), cap mix A (5% phenoxyacetic anhydride (Pac2O) in tetrahydrofuran (THF)/pyridine), cap mix B (16% methylimidazole/THF), oxidizing solution (iodine (I2) 0.02 M in water/pyridine/THF), deblocking mix (3% trichloroacetic acid (TCA) in dichloromethane), deprotection solution (0.05 M potassium carbonate (K2CO3) in methanol (MeOH)), and triethylammonium acetate 2 M (TEAA) were purchased from Glen Research (Sterling, Virginia). Acetonitrile (CH3CN, DNA synthesis grade) and dichloromethane (DNA synthesis grade) were purchased from Biosolve (Valkenswaard, Netherlands). All reagents were used without further purification. Complementary HBV DNA and non-complementary (23mer polyThymidine) were purchased from Eurogentec (Angers, France).
CPG (200–400 Mesh, 300 nm average pore size) was purchased from Fluka. Fluorescent rhodamine B NPs (diameter ∼50 nm) were provided by Nano-H (Lyon, France). The concentration of the NP solution was 9.3 × 1013 ± 9 × 1012 NP mL−1. This reference concentration was calculated by first measuring the amount of silicon in the solution (5.0 ± 0.5 g L−1 silicon) by Inductively Coupled Plasma Atomic Emission Spectroscopy (ICP-AES) and then taking a mean particle diameter of 50 nm and a mass density of 1.75 g cm−3 for calculation. Indeed, colloidal organo-silica spheres (i.e. organic molecules like APTES and rhodamine embedded in silica core) have a lower mass density than silica (1.96 g cm−3).30
Measurements
1H NMR spectra were recorded on a Bruker DRX-300 using deuterated chloroform as a solvent. Scanning electron microscopy (SEM) pictures were obtained using a Hitachi S800 FEG with an accelerating voltage of 10–15 kV (Centre Technologique des Microstructures - Lyon 1, Villeurbanne, France). SEM samples were metallized in sputtering mode with a 4 nm layer of Au/Pd. Transmission electron microscopy (TEM) pictures were obtained using a Philips CM120 with an accelerating voltage of 120 kV. Silica NPs were examined after deposition of 5 μL of diluted solutions on a formvar-coated copper grid (Electron Microscopy Sciences) and evaporation to dryness. Optical microscopy fluorescence images were taken using Zeiss Axioplan 2 Imaging apparatus, equipped with ×10 and ×40 lenses and a monochrome camera. Samples were observed by both fluorescence and transmitted light. Rhodamine was excited by a lamp with a 550 (± 25) nm band-pass filter and fluorescence from the sample was collected with a 605 (± 70) nm band-pass filter. Dynamic Light Scattering (DLS) measurements were carried out with a Zetasizer NanoZS (Malvern Instruments) to determine the hydrodynamic diameter and polydispersity index (PdI) of the NP. Concentrations of NP solutions were determined by Inductively Coupled Plasma Atomic Emission Spectroscopy (ICP-AES) with a Thermo Scientific iCAP 6300. Oligonucleotides were synthesized using an Applied Biosystems 394 RNA/DNA synthesizer (Applied Biosystems, Foster City, CA) using the standard 1 μmol synthesis program for phosphoramidite chemistry. Absorbance measurements of dimethoxytrityl cations (DMT) and oligonucleotides were performed on a Varian Cary 100 Bio UV-Visible spectrophotometer (Agilent Technologies, Santa Clara, CA) using a quartz cuvette of 1 cm path length. Absorbance at 498 nm and 260 nm was measured for the DMT group (ε = 70 mL μmol−1 cm−1) and oligonucleotide quantifications, respectively. MALDI-ToF mass spectrometry analysis was performed using a Voyager DE-PRO Applied Biosystems instrument with 3-hydroxypicolinic acid as the matrix (IBCP, Lyon, France). Analysis of oligonucleotide purity was performed with an Agilent 1200 series High Performance Liquid Chromatography (HPLC) on a Licrospher RP18 column with 1 mL min−1 flow rate of the mobile phase. Gradient elution was from 4.5 to 25% of acetonitrile in a triethylammonium acetate (TEAA) buffer 0.05 M over 30 min. Time-of-Flight Secondary Ion Mass Spectrometry (ToF-SIMS) measurements were carried out using a Physical Electronics TRIFT III ToF-SIMS instrument operated with a pulsed 22 keV Au+ ion gun (ion current of 2 nA) rastered over 300 μm × 300 μm. An electron gun was operated in pulsed mode at low electron energy to allow charge compensation. Ion dose was kept below the static conditions limit. Data were analysed using WinCadence software. Mass calibration was performed on hydrocarbon secondary ions. Each sample was pressed on indium foil prior to analysis. Atomic Force Microscopy was performed using a semi-contact mode AFM (NT-MDT, Eindhoven, Netherlands).Methods
Synthesis of 3-(triethoxysilyl)propyl-hydroxyhexyl urea. The synthesis of 3-(triethoxysilyl)propyl-hydroxyhexyl urea (TESPHU) was achieved as described in a previous paper.11 6-Amino-1-hexanol (1.25 mmol, 146 mg) was first dissolved in 2.5 mL of isopropanol. After the addition of 3-(triethoxysilyl)propyl isocyanate (1.25 mmol, 310 μL), the mixture was stirred at room temperature for 30 min. An aliquot of this solution was evaporated to dryness under reduced pressure and characterized by NMR.
1H NMR (300 MHz, CDCl3): δ (ppm) = 0.59 (t, 2H, Si–CH2), 1.19 (t, 9H, 3CH3), 1.35–1.60 (m, 10H, 5CH2), 3.10–3.20 (m, 4H, 2CH2N), 3.64 (t, 2H, CH2–OH), 3.76–3.87 (q, 6H, 3CH2OSi), 4.82–4.96 (2t, 2H, 2NH).
Silica nanoparticle silanization. A crude solution of TESPHU (563 μL, 0.5 M) was added to a suspension of silica NPs (200 mg) in 20 mL of DMF, the mixture was slowly stirred at 120 °C for 16 h. The suspension was centrifuged (10
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g, 45 min), and the supernatant was eliminated. Then, the particles were washed during three centrifugation/redispersion cycles in DMF. For each wash cycle, sonication was used to redisperse the TESPHU-NP completely. Silica NPs and TESPHU-NP were measured to be 51.7 ± 8.3 nm and 52.9 ± 7.2 nm respectively, using TEM (average of at least 120 measurements on ImageJ software).
000 g, 45 min), and the supernatant was eliminated. Then, the particles were washed during three centrifugation/redispersion cycles in DMF. For each wash cycle, sonication was used to redisperse the TESPHU-NP completely. Silica NPs and TESPHU-NP were measured to be 51.7 ± 8.3 nm and 52.9 ± 7.2 nm respectively, using TEM (average of at least 120 measurements on ImageJ software).
Amine functionalization. Controlled pore glass particles (2 g) were suspended in concentrated nitric acid (10 mL) in a round-bottom flask to activate the silanols. The suspension was heated under reflux for 1 h. Then, the CPG particles were washed with distilled water in a fritted-glass filter until neutral pH.
A 10% (v/v) aqueous solution of 3-aminopropyltriethoxysilane (APTES) was prepared and the pH was adjusted to 4 with hydrochloric acid. Activated CPG particles were gently agitated in 15 mL of APTES solution at 70 °C for 2.5 h. The excess solution was removed and the wet beads were placed in an oven at 120 °C overnight to polymerize the silane. The CPG particles were rinsed in a fritted-glass filter with acetone (10 mL, 3 times) and dried.
Carboxylic acid functionalization. The CPG-NH2 beads (2 g) were suspended in 15 mL of a solution containing 1% (v/v) of triethylamine and 10% (v/v) of succinic anhydride in acetone. The mixture was gently agitated at room temperature for 5 h. The CPG particles were rinsed in a fritted-glass filter with acetone (10 mL, 3 times) and air-dried.
CPG activation. The CPG-COOH beads (2 g) were suspended in 20 mL of a solution containing HBTU (114 mg), DMAP (37 mg) and triethylamine (42 μL) dissolved in anhydrous acetonitrile (20 mL). The mixture was gently agitated at room temperature for 16 h. CPG particles were rinsed in fritted-glass filter with dimethylformamide (20 mL, 3 times), then with dichloromethane (20 mL, 3 times) and dried.
NP grafting. CPG particles with activated acids were suspended in DMF (8 mL) in a round-bottom flask. TESPHU-NP solution (200 mg in 20 mL DMF) was added dropwise under agitation. The suspension was stirred at room temperature for at least 2 h. The NP-CPG assemblies were rinsed in a fritted-glass filter with DMF (20 mL, 3 times) to remove non-grafted NP.
Capping step. Non-reacting activated acids were capped in piperidine (10 mL) for 6 days at room temperature. NP-CPG assemblies were rinsed in a fritted-glass filter with DMF (20 mL, 3 times), then with anhydrous acetonitrile (20 mL, 3 times) and dried.
Quantification of grafted NP. After the grafting reaction, the residual NP solution was transferred to a 25 mL volumetric flask which was then filled with DMF. Absorbance at 554 nm was measured in a 1 mL quartz cuvette. The value was corrected with a blank sample. The calibration curve was plotted using TESPHU-NP solutions at different concentrations in DMF (Fig. S3 in the ESI†).
Oligonucleotides were synthesized using a standard protocol (1 μmol scale coupling program). Acetonitrile washing steps were increased for efficient washing of support during synthesis. ∼35–40 mg of NP-CPG was introduced in each column. After the first and last nucleotide incorporations, DMT groups were released from the support with a deblocking mix solution and titrated at 498 nm (ε = 70 ml μmol−1 cm−1). The stability of the assemblies after DNA synthesis was controlled by SEM. Nucleic bases were then deprotected for 16 h in 1 mL K2CO3 0.05 M in methanol; no NP release was observed during this step.
The quality of the DNA sequence synthesized from the assemblies was controlled by HPLC and MALDI-ToF mass spectrometry. To do so, it was necessary to synthesize a DNA which could be released from the nanoparticles. A specific base-cleavable sulfone derivative (chemical phosphorylation reagent) was coupled on the NPs prior to initiating HBV DNA synthesis. This derivative was cleaved during the deprotection step (6 h in 1 mL K2CO3 0.05 M in methanol) to liberate an HBV DNA-3′-phosphate in solution (ATCTCGGGAATCTCAATGTTAGT-(PEG)1-PO32−). After filtration to remove the support, the solution was neutralized with 1.5 mL triethylammonium acetate 2 M, then concentrated to 500 μL using a 3K Amicon Ultra filter (Merck Millipore, MA, USA). DNA quality was determined by HPLC, then concentrated to dryness and analysed by MALDI-ToF MS ([M + H]+ calculated 7344.6; found 7344.3).
Grafting onto SPRi chips. For the modification of the SPRi chips with oligonucleotide probes, the diazonium technology developed by the authors was used.33 Briefly, the surface was modified by an electro-grafted carboxymethyl aniline (CMA), and subsequently activated to generate a covalent linkage with amine terminated oligonucleotide.
Prior to its electro-deposition, the CMA was diazotized in order to generate the necessary aryl diazonium function. For this purpose, a solution of 20 mM ice-cold CMA was incubated for 20 min in the presence of 15 mM HCl and 15 mM NaNO2. The diazonium solution formed was then immediately used to perform electro-addressing by directly depositing a 100 μL drop on the surface of the SPRi chip. Three cyclic voltammograms were then performed, from 0 to −2 V vs. Pt, at a scan rate of 200 mV s−1. The cyclic voltammetry experiments were conducted using a VMP3 potentiostat (BioLogic, France).
Following surface modification, the SPRi chips were rinsed with distilled water, air dried and incubated for 90 min in an activating solution composed of 0.1 M EDC, 0.1 M NHS. At the end of this step, the chips were ready for probe immobilization. In order to generate probe arrays, 0.9 nL drops of DNA solutions were deposited using a sciFLEXARRAYER S3 (SCIENION, Germany) non-contact piezoelectric spotter and incubated for 30 min at 37 °C in a humidity saturated chamber. Two different plots were prepared. The positive plot was coated with the complementary oligonucleotide of the 23mer HBV DNA (5′-CTAACATTGAGATTCCCGAGATT-3′) and the negative plot was coated with a 23mer polyThymidine. The chip surface was then extensively washed with water and saturated for 20 min at 37 °C with PBSTA (100 mM PBS, pH 7.4 with addition of 0.1% Tween 20 and 1% BSA).
SPRi interaction monitoring. All interaction studies were performed in 100 mM PBS buffer, pH 7.4 on a SPRi-biochip pre-saturated with PBS containing BSA 1%, Tween 20 0.1%, and using a SPRi-lab+ (Genoptics, France). The samples were injected in the 6 μL flow cell using a 500 μL injection loop. Two different dilutions of the DNA-NP solution were tested (dilution 1/100 = 6 × 1012 NP mL−1 and dilution 1/25 = 2.4 × 1013 NP mL−1). A constant 130 μL min−1 flow rate was maintained during the entire experiment using a peristaltic pump.
Conclusions
NP-CPG silica micro-sized assemblies were elaborated in order to achieve DNA synthesis onto nano-sized particles directly in a DNA synthesizer, using phosphoramidite chemistry. The surface within the pores of the CPG material appeared to be covered by a homogeneous layer of NPs. We demonstrated that the pores protected NP anchoring during the DNA synthesis without decreasing the coupling yield of the phosphoramidite synthons. The DNA strands synthesized on the NP surface were found to be of good quality and accessible for binding with complementary Hepatitis B Virus (HBV) DNA. Our bottom-up strategy for NP functionalization with DNA led to unprecedented DNA loading efficiency.Acknowledgements
The authors would like to thank the ANR (ANR-08-Biotecs-003), LyonBiopole and Lyon Science Transfert for their financial support of the project.References
- J. Yan, M. C. Estévez, J. E. Smith, K. Wang, X. He, L. Wang and W. Tan, Nano Today, 2007, 2, 44–50 CrossRef.
- J. E. Smith, L. Wang and W. Tan, TrAC, Trends Anal. Chem., 2006, 25, 848–855 CrossRef CAS.
- R. A. Petros and J. M. DeSimone, Nat. Rev. Drug Discovery, 2010, 9, 615–627 CrossRef CAS.
- S. Jin and K. Ye, Biotechnol. Prog., 2007, 23, 32–41 CrossRef CAS.
- X. Zhao, R. Tapec-Dytioco and W. Tan, J. Am. Chem. Soc., 2003, 125, 11474–11475 CrossRef CAS.
- D. Knopp, D. Tang and R. Niessner, Anal. Chim. Acta, 2009, 647, 14–30 CrossRef CAS.
- J.-L. Bridot, A.-C. Faure, S. Laurent, C. Rivière, C. Billotey, B. Hiba, M. Janier, V. Josserand, J.-L. Coll, L. Vander Elst, R. Muller, S. Roux, P. Perriat and O. Tillement, J. Am. Chem. Soc., 2007, 129, 5076–5084 CrossRef CAS.
- N. Geerts and E. Eiser, Soft Matter, 2010, 6, 4647–4660 RSC.
- S. Jiang, M. K. Gnanasammandhan and Y. Zhang, J. R. Soc. Interface, 2009, 7, 3–18 CrossRef.
- S. Park and K. Hamad-Schifferli, Curr. Opin. Chem. Biol., 2010, 14, 616–622 CrossRef CAS.
- C. Farre, M. Lansalot, R. Bazzi, S. Roux, C. A. Marquette, G. Catanante, L. J. Blum, N. Charvet, C. Louis and C. Chaix, Langmuir, 2010, 26, 4941–4950 CrossRef CAS.
- H. Seliger, M. Hinz, R. Ditz, M. Koch, P. Lapido and S. Margel, Nucleosides, Nucleotides Nucleic Acids, 2007, 26, 1167–1172 CAS.
- D. S. Seferos, A. E. Prigodich, D. A. Giljohann, P. C. Patel and C. A. Mirkin, Nano Lett., 2009, 9, 308–311 CrossRef CAS.
- D. A. Giljohann, D. S. Seferos, P. C. Patel, J. E. Millstone, N. L. Rosi and C. A. Mirkin, Nano Lett., 2007, 7, 3818–3821 CrossRef CAS.
- R. T. Pon, inCurrent Protocols in Nucleic Acid Chemistry, John Wiley & Sons, Inc., 2001 Search PubMed.
- S. L. Beaucage and R. P. Iyer, Tetrahedron, 1992, 48, 2223–2311 CrossRef CAS.
- C. J. May, H. E. Canavan and D. G. Castner, Anal. Chem., 2004, 76, 1114–1122 CrossRef CAS.
- H. D. Hill, J. E. Millstone, M. J. Banholzer and C. A. Mirkin, ACS Nano, 2009, 3, 418–424 CrossRef CAS.
- D. Giaume, M. l. Poggi, D. Casanova, G. v. Mialon, K. Lahlil, A. Alexandrou, T. Gacoin and J.-P. Boilot, Langmuir, 2008, 24, 11018–11026 CrossRef CAS.
- Y. Chen and Y. Zhang, Anal. Bioanal. Chem., 2011, 399, 2503–2509 CrossRef CAS.
- T. Schiestel, H. Brunner and G. E. M. Tovar, J. Nanosci. Nanotechnol., 2004, 4, 504–511 CrossRef CAS.
- S. J. Hurst, A. K. R. Lytton-Jean and C. A. Mirkin, Anal. Chem., 2006, 78, 8313–8318 CrossRef CAS.
- Y. Wang, K. Y. Pu and B. Liu, Langmuir, 2010, 26, 10025–10030 CrossRef CAS.
- J. Wu, J. Silvent, T. Coradin and C. Aime, Langmuir, 2012, 28, 2156–2165 CrossRef CAS.
- K. S. Rao, S. U. Rani, D. K. Charyulu, K. N. Kumar, B. K. Lee, H. Y. Lee and T. Kawai, Anal. Chim. Acta, 2006, 576, 177–183 CrossRef CAS.
- W. Zhao, W. Chiuman, J. C. F. Lam, S. A. McManus, W. Chen, Y. Cui, R. Pelton, M. A. Brook and Y. Li, J. Am. Chem. Soc., 2008, 130, 3610–3618 CrossRef CAS.
- M. R. Jones, R. J. Macfarlane, A. E. Prigodich, P. C. Patel and C. A. Mirkin, J. Am. Chem. Soc., 2011, 133, 18865–18869 CrossRef CAS.
- J. A. Dougan, C. Karlsson, W. E. Smith and D. Graham, Nucleic Acids Res., 2007, 35, 3668–3675 CrossRef CAS.
- Z. Li, T. Niu, Z. Zhang, R. Chen, G. Feng and S. Bi, Biosens. Bioelectron., 2011, 26, 4564–4570 CrossRef CAS.
- A. van Blaaderen and A. Vrij, J. Colloid Interface Sci., 1993, 156, 1–18 CrossRef CAS.
- B. de Lambert, C. Chaix, M. T. Charreyre, T. Martin, A. Aigoui, A. Perrin-Rubens, C. Pichot and B. Mandrand, Anal. Biochem., 2008, 373, 229–238 CrossRef CAS.
- C. Minard-Basquin, C. Chaix, C. Pichot and B. Mandrand, Bioconjugate Chem., 2000, 11, 795–804 CrossRef CAS.
- C. A. Mandon, L. J. Blum and C. A. Marquette, ChemPhysChem, 2009, 10, 3273–3277 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: Additional SEM and optical microscopy images of NP-CPG assemblies; Absorbance spectra and calibration curve of the reference NP solution; Stability study of NPs in ammonia by TEM; ToF-SIMS spectra of silica NP, TESPHU-NP and DNA-NP; MALDI-ToF mass spectrometry of the synthetic DNA and Dynamic Light Scattering of NPs. See DOI: 10.1039/c2ra22077f |
| This journal is © The Royal Society of Chemistry 2012 |