Ti(III)-Catalyzed, concise synthesis of marine furanospongian diterpenes†
Antonio
Rosales
*ab,
Juan
Muñoz-Bascón
a,
Víctor
Manuel Morales-Alcázar
a,
José A.
Castilla-Alcalá
b and
J.
Enrique Oltra
*a
aDepartment of Organic Chemistry, Faculty of Sciences, University of Granada, 18071, Granada, Spain. E-mail: joltra@ugr.es; Fax: +34 958248437
bDepartment of Clinic Analysis, University Hospital Virgen de las Nieves, 18014, Granada, Spain. E-mail: a.rosales.martinez@gmail.com
First published on 26th October 2012
Abstract
A bioinspired procedure for the straightforward synthesis of marine furanospongian diterpenes is described. The key step is the titanocene(III)-catalyzed radical cascade cyclization of the suitable epoxy-polyprene.
Introduction
In recent years radical cyclizations of epoxypolyenes catalyzed by Cp2TiCl (Nugent's reagent)1–4 have become powerful tools in organic synthesis.5–8 In fact this reaction has facilitated considerably the straightforward synthesis of several cyclic terpenes, including monoterpenoids, such as karahanaenone;9 sesquiterpenoids, such as trans-4(11),8-daucadiene,9 isodrimenediol,10 3β-hydroxydihydroconfertifolin,10 tuberiferin,11 and dehydrobrachylaenolide;11 diterpenoids, such as 3β-hydroxymanool,12 rostratone,13,14 aphidicolin,14 pyripyropene A,14 barekoxide,9 and laukarlaool;9 triterpenoids, such as 3β-hydroxymalabaricatriene12 and achilleol A;15 meroterpenoids, such as stypoldione,12 zonarone16 and zonarol;16 and the ambergris-type odorant α-ambrinol.17 We have extended the Ti(III)-catalyzed radical cascade cyclization to the synthesis of furanospongian diterpenes, thus reinforcing the hypothesis that with a judicious choice of epoxy-polyprene starting material this method allows the straightforward synthesis of terpenoids with many different skeletons.Results and discussion
Spongian diterpenes constitute a subfamily of marine terpenoids with a spongian skeleton (1)18 among which furanospongian diterpenes such as 2 and 3 incorporate a heteroaromatic furan D ring.18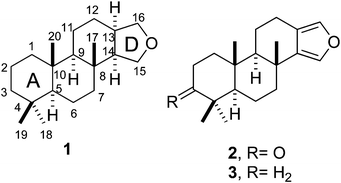 | ||
| Chart 1 Spongian skeleton (1) and chemical structure of furanospongian diterpenes 2 and 3. | ||
Furanoditerpenoids 2 and 3 were first isolated from the nudibranch Glossodoris atromarginata19 and the sponge Spongia officinalis,20 respectively, and have proved to exert a cytotoxic effect on tumor cells,21 antiviral activity,21 and an inhibitory effect on the development of sea-urchin embryos.22 These terpenoids are scarce in nature, however, and harvesting their biological sources from the sea is no easy task. Within this context, chemical synthesis may provide additional supplies to facilitate their pharmacological study. In fact, the total synthesis of 2 and 3 was reported as long ago as 1995,23 and four years later both compounds were prepared from S−(+)-carvone.24 Nevertheless, the total synthesis of these products requires more than twenty steps23,25 and twelve or thirteen for preparation from (+)-carvone,24 and even then affords low overall yields.
It is well known that the biosynthesis of steroids derives from the enzyme-catalyzed cyclization of 2,3-epoxy-squalene.26 Inspired by this biosynthesis, we deemed that the synthesis of furanospongian diterpenoid 2 could be efficiently achieved by means of the key titanocene(III)-catalyzed cascade cyclization of epoxy-geranylgeraniol (5) (Scheme 1). Eventually, ketone 2 would be reduced to 1.
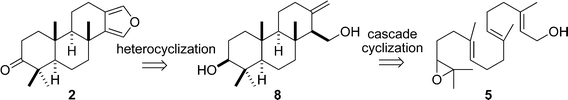 | ||
| Scheme 1 Bioinspired retrosynthetic analysis of 2. | ||
As expected, the titanocene(III)-catalyzed cascade cyclization of racemic epoxy-geranylgeraniol derivative 6, the starting material for our synthesis of stypoldione,12 gave a 36% yield of tricyclic olefin 7 bearing the crucial exocyclic double bond (Scheme 2). This yield should be regarded as entirely satisfactory if we bear in mind that this Ti(III)-catalyzed cyclization selectively afforded a product containing three fused (trans/anti/trans) six-membered rings, an exocyclic alkene, and six stereogenic centers out of 192 possible regio- and stereoisomers.
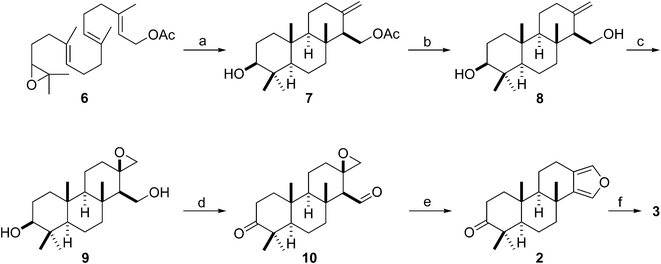 | ||
| Scheme 2 Synthesis of 2 and 3 from epoxy-polyprene 6. (a) Cp2TiCl2 (0.2 equiv.), Mn (8 equiv.), Me3SiCl (4 equiv.), 2,4,6-collidine (7 equiv.), THF, rt, 12 h, 36%; (b) K2CO3, MeOH, 5 °C, 3 h, 85%; (c) MCPBA, DCM, 5 °C, 2 h, 95%; (d) Dess–Martin periodinane, rt, 5 h, 99%; (e) p-TsOH; DCM–DMSO, 50 °C, 6 h, 73%; (f) ref. 10, 1 step, 75%. | ||
Simple saponification of acetate 7 unmasked the primary alcohol group of 8, which directed the following epoxidation reaction to give stereoselectively a 95% yield of β-epoxide 9 (stereochemistry of the oxirane ring was tentatively assigned due to the β-disposition of the hydroxyl-methyl directing group). Dess–Martin oxidation of diol 9 afforded an almost 100% yield of ketoaldehyde 10, which already had in place the epoxide and carbonyl functions required for the next heterocyclization step. Subsequently, the acidic treatment of 10 provided a 73% yield of synthetic furanospongian diterpene 2, the NMR data of which matched those reported for the natural product isolated from G. atromarginata.19 Thus, the synthesis of 2 from epoxy-polyene 6 was completed in only five steps to afford a 21% overall yield, substantially improving on the synthetic procedures mentioned above.23,24 Moreover, the reduction of ketone 2 to diterpene 3 has already been reported,24 and so Scheme 2 also represents the formal synthesis of the metabolite from the sponge S. officinalis (3) in six steps. Bearing in mind that titanium is one of the most abundant safe transition metals on Earth,27 our results suggest that this titanocene(III)-catalyzed procedure might become a general method for the synthesis of furanospongian diterpenes.
Experimental
General methods
The general details have been described elsewhere.28 Silica gel was used as solid support for flash chromatography. Compounds 2,196,12 and 712 are known and their NMR spectra matched those previously reported.Cp2TiCl-catalyzed cyclization of epoxy-polyprene 6
Strictly deoxygenated THF (4 mL) was added to a mixture of Cp2TiCl2 (21 mg, 0.086 mmol) and Mn dust (188 mg, 3.44 mmol) under an Ar atmosphere and the suspension was stirred at room temperature until it turned lime green (after about 15 min). Then, a solution of epoxy-polyprene 6 (150 mg, 0.43 mmol) and 2,4,6-collidine (0.36 mL, 3.0 mmol) in THF (0.5 mL), and Me3SiCl (0.21 mL, 1.72 mmol) were added and the solution was stirred for 12 h. The reaction was then quenched with 2N HCl and extracted with Et2O. The organic layer was washed with brine, dried (anhyd. Na2SO4), and the solvent removed. The residue was dissolved in THF (4 mL) and stirred with Bu4NF (350 mg, 1.72 mmol) for 3 h. The mixture was then diluted with Et2O, washed with brine, dried (anhyd. Na2SO4), and the solvent removed. The residue was submitted to flash chromatography (hexane–AcOEt 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) affording 7 (54 mg, 36% yield). The NMR data of 7 matched those previously reported.12
:2) affording 7 (54 mg, 36% yield). The NMR data of 7 matched those previously reported.12
Preparation of diol 8
A solution of compound 7 (38 mg, 0.110 mmol) in MeOH (1 mL) was cooled to 5 °C and K2CO3 (95 mg, 0.69 mmol) was added. The reaction mixture was stirred for 3 h, the solvent was then removed and the residue was diluted in Et2O and washed with brine. The organic layer was dried (anhyd. Na2SO4) and the solvent removed. The residue was submitted to flash chromatography (hexane–EtOAc, 7:3) to give diol 8 (29 mg, 85%) as an amorphous solid. IR (film) νmax cm−1 = 3337, 2850, 1644, 1442; 1H NMR (500 MHz, CDCl3): δ = 4.93 (d, J = 1.0 Hz, 1 H-16a), 4.64 (br d, J = 1.0 Hz, 1 H-16b), 3.82 (dd, J = 11.0, 3.8 Hz, 1 H-15a), 3.78 (dd, J = 10.9, 9.4 Hz, 1 H-3), 3.22 (dd, J = 11.6, 4.8 Hz, 1 H-15b), 3.15–3.09 (m, 1H), 2.41 (ddd, J = 12.8, 4.2, 2.4 Hz, 1 H), 1.95 (dd, J = 9.7, 3.4 Hz, 1 H), 1.78 (dt, J = 12.5, 3.1 Hz, 1 H), 1.73–1.20 (m, 11 H), 0.99 (s, 3H), 0.81 (s, 3H), 0.77 (s, 3 H), 0.72 (s, 3 H); 13C NMR (126 MHz, CDCl3, DEPT): δ = 147.5 (C), 106.2 (CH2), 78.8 (CH), 59.7 (CH), 59.4 (CH), 58.7 (CH2), 55.3 (CH), 40.6 (CH2), 39.1 (C), 38.8 (C), 38.5 (CH2), 37.6 (CH2), 37.5 (C), 29.7 (CH2), 28.3 (CH3), 27.3 (CH2), 18.6 (CH2), 16.3 (CH3), 16.2 (CH3), 15.3 ppm (CH3); HRMS (FAB): calcd. for C20H34NaO2 [M + Na]+ 329.2457, found 329.2478.
Synthesis of epoxide 9
A solution of diol 8 (25 mg, 0.081 mmol) in CH2Cl2 (5 mL) was cooled to 5 °C and MCPBA (70%) (25 mg, 0.1 mmol) was added. The reaction mixture was stirred for 2 h and then diluted with Et2O. The ethereal solution was washed with 0.5 M Na2S2O4, saturated aqueous Na2CO3 and brine, dried (anhyd. Na2SO4) and the solvent was removed. The residue was submitted to flash chromatography (hexane–EtOAC, 95:5) to give epoxide 9 (24 mg, 95%) as an amorphous solid. IR (film) νmax cm−1 = 3510, 2850 1213; 1H NMR (500 MHz, CDCl3): δ = 3.63 (dd, J = 11.6, 3.3 Hz, 1 H-15a), 3.43 (dd, J = 11.3, 10.4 Hz, 1 H-3), 3.24 (dd, J = 11.6, 4.7 Hz, 1 H-15b), 3.21 (dd, J = 3.7, 2.1 Hz, 1 H-16a), 2.72 (d, J = 3.7 Hz, 1 H-16b), 2.00–1.93 (m, 1 H), 1.88 (dd, J = 10.2, 3.2, 1 H), 1.83–1.78 (m, 2 H), 1.76–1.20 (m, 11 H), 1.00 (s, 3 H), 0.86 (s, 3 H), 0.85 (s, 3 H), 0.79 (s, 3H); 13C NMR (126 MHz, CDCl3) δ = 78.7 (CH), 59.1 (CH), 58.8 (CH2), 57.9 (C), 55.2 (CH), 54.6 (CH), 51.7 (CH2), 40.5 (CH2), 39.3 (C), 38.8 (C), 38.5 (CH2), 37.4 (C), 36.1 (CH2), 29.7 (CH3), 28.0 (CH2), 27.2 (CH2), 18.2 (CH2), 16.6 (CH3), 16.3 (CH3), 15.3 (CH3) ppm. HRMS (FAB): calcd. for C20H34NaO3 [M + Na]+ 345.2406, found 345.2425.
Synthesis of aldehyde 10
Dess–Martin periodinane (58 mg, 0.136 mmol) was added to a solution of epoxide 9 (20 mg, 0.062 mmol) in CH2Cl2 (3 mL) at room temperature. The reaction mixture was stirred for 5 h and then diluted with CH2Cl2. The CH2Cl2 solution was washed with a saturated solution of a 1:1 mixture of NaHCO3 and Na2SO3 and with brine. After drying and removal of the solvent the residue was submitted to flash chromatography (hexane–EtOAc, 95:5) to give aldehyde 10 (19 mg, quant.) as an amorphous solid. IR (film) νmax cm−1 = 2851, 1737, 1706; 1H NMR (500 MHz, CDCl3) δ = 9.58 (d, J = 3.4 Hz, 1H-15), 3.15–3.10 (m, 1H-16a), 2.72 (d, J = 3.3 Hz, 1H-16b), 2.54–2.44 (m, 3H), 2.0–1.90 (m, 2H), 1.85 (dt, J = 12.8, 2.9 Hz, 1H), 1.75–1.5 (m, 9H), 1.24 (s, 3H), 1.11 (s, 3H), 1.05 (s, 3H), 0.98 (s, 3H) ; 13C NMR (151 MHz, CDCl3, DEPT) δ 219.7 (C), 205.0 (CH), 66.9 (CH), 60.4 (CH), 57.3 (CH), 54.7 (CH2), 49.9 (C), 43.2 (C), 42.3 (CH2), 41.7 (CH2), 39.7 (C), 38.2 (CH2), 36.5 (CH2), 27.1 (C), 23.5 (CH2), 21.7 (CH2), 18.8 (CH3), 16.8 (CH3), 11.3 ppm (2CH3); HRMS (FAB): m/z calcd. for C20H30O3Na [M + Na]+: 341.2093; found: 341.2110.
Synthesis of furanospongian diterpene 2
To a solution of aldehyde 10 (15 mg, 0.05 mmol) in CH2Cl2 (0.18 mL) and DMSO (0.27 mL) a solution of anhydrous PTSA in DMSO (0.27 mL, 330 mg p-TsOH mL−1) was added. The reaction mixture was warmed to 50 °C and stirred for 6 h. The reaction mixture was then diluted with diethyl ether and washed with brine. The organic layer was dried over Na2SO4 and the solvent removed. The residue was submitted to flash chromatography to give furanospongian diterpene 2 (11 mg, 73%). The NMR data of synthetic 2 matched those previously reported for the natural product.19Conclusion
Here we describe a novel procedure for the straightforward synthesis of bioactive spongian diterpenes 2 and 3. The key step is the titanocene(III)-catalyzed radical cascade cyclization of epoxy-polyprene 6.Acknowledgements
This work was financed by the Spanish MICINN (Project CTQ2011-24443) and the Regional Government of Andalucía (Projects P07-FQM-3213 and P10-FQM-6050). The authors thank their English colleague Dr Jon Trout for revising their English text.References
- T. V. RajanBabu and W. A. Nugent, J. Am. Chem. Soc., 1994, 116, 986–997 CrossRef CAS , and references therein.
- A. Gansäuer, H. Bluhm and M. Pierobon, J. Am. Chem. Soc., 1998, 120, 12849–12859 CrossRef , and references therein.
- A. F. Barrero, A. Rosales, J. M. Cuerva and J. E. Oltra, Org. Lett., 2003, 5, 1935–1938 CrossRef CAS.
- A. Gansäuer, C. A. Fan, J. Justicia and D. Worgull, Top. Curr. Chem., 2007, 279, 25–52 CrossRef.
- J. M. Cuerva, J. Justicia, J. L. Oller-López and J. E. Oltra, Top. Curr. Chem., 2006, 264, 63–91 CrossRef CAS.
- J. M. Cuerva, J. Justicia, J. L. Oller-López, B. Bazdi and J. E. Oltra, Mini-Rev. Org. Chem., 2006, 3, 23–35 Search PubMed.
- J. Justicia, L. Álvarez de Cienfuegos, A. G. Campaña, D. Miguel, V. Jakoby, A. Gansäuer and J. M. Cuerva, Chem. Soc. Rev., 2011, 40, 3525–3537 RSC.
- A. Gansäuer, L. Shi, M. Otte, I. Huth, A. Rosales, I. Sancho-Sanz, N. M. Padial and J. E. Oltra, Top. Curr. Chem., 2012, 320, 93–120 Search PubMed.
- J. Justicia, J. L. Oller-López, A. G. Campaña, J. E. Oltra, J. M. Cuerva, E. Buñuel and D. J. Cárdenas, J. Am. Chem. Soc., 2005, 127, 14911–14921 CrossRef CAS.
- J. Justicia, J. E. Oltra, A. F. Barrero, A. Guadaño, A. González-Coloma and J. M. Cuerva, Eur. J. Org. Chem., 2005, 712–718 CrossRef CAS.
- J. Justicia, L. Álvarez de Cienfuegos, R. E. Estévez, M. Paradas, A. M. Lasanta, J. L. Oller, A. Rosales, J. M. Cuerva and J. E. Oltra, Tetrahedron, 2008, 64, 11938–11943 CrossRef CAS.
- J. Justicia, A. Rosales, E. Buñuel, J. L. Oller-López, M. Valdivia, A. Haïdour, J. E. Oltra, A. F. Barrero, D. J. Cárdenas and J. M. Cuerva, Chem.–Eur. J., 2004, 10, 1778–1788 CrossRef CAS.
- J. Justicia, J. E. Oltra and J. M. Cuerva, Tetrahedron Lett., 2004, 45, 4293–4296 CrossRef.
- J. Justicia, J. E. Oltra and J. M. Cuerva, J. Org. Chem., 2005, 70, 8265–8270 CrossRef CAS.
- A. F. Barrero, J. M. Cuerva, E. J. Álvarez-Manzaneda, J. E. Oltra and R. Chahboun, Tetrahedron Lett., 2002, 43, 2793–2796 CrossRef CAS.
- A. Gansäuer, J. Justicia, A. Rosales, D. Worgull, B. Rinker, J. M. Cuerva and J. E. Oltra, Eur. J. Org. Chem., 2006, 4115–4127 CrossRef.
- J. Justicia, A. G. Campaña, B. Bazdi, R. Robles, J. M. Cuerva and J. E. Oltra, Adv. Synth. Catal., 2008, 350, 571–576 CrossRef CAS.
- R. A. Keyrers, P. T. Northcote and M. T. Davies-Coleman, Nat. Prod. Rep., 2006, 23, 321–334 RSC.
- M. J. Somerville, E. Mollo, G. Cimino, W. Rungprom and M. J. Garson, J. Nat. Prod., 2006, 69, 1086–1088 CrossRef CAS.
- N. Capelle, J. C. Braekman, D. Daloze and B. Tursch, Bull. Soc. Chim. Belg., 1980, 89, 399–404 CAS.
- L. Betancur-Galvis, C. Zuluaga, M. Arnó, M. A. Gonzáles and R. J. Zaragozá, J. Nat. Prod., 2002, 65, 189–192 CrossRef CAS.
- L. P. Ponomarenko, N. A. Terent'eva, V. B. Krasokin, A. I. Kalinovsky and V. A. Rasskazov, Nat. Prod. Commun., 2011, 6, 773–776 Search PubMed.
- T. Sakamoto and K. Kanematsu, Tetrahedron, 1995, 51, 5771–5780 CrossRef CAS.
- M. Arnó, M. A. Gonzalez and R. J. Zaragozá, Tetrahedron, 1999, 55, 12419–12428 CrossRef CAS.
- Y. Baba, T. Sakamoto, S. Soejima and K. Kanematsu, Tetrahedron, 1994, 50, 5645–5658 CrossRef CAS.
- R. A. Yoder and J. N. Johnston, Chem. Rev., 2005, 105, 4730–4756 CrossRef CAS.
- D. J. Ramón and M. Yus, Chem. Rev., 2006, 106, 2126–2208 CrossRef CAS.
- M. Paradas, A. G. Campaña, T. Jiménez, R. Robles, J. E. Oltra, E. Buñuel, J. Justicia, D. J. Cárdenas and J. M. Cuerva, J. Am. Chem. Soc., 2010, 132, 12748–12756 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra22281g |
| This journal is © The Royal Society of Chemistry 2012 |