DOI:
10.1039/C2RA21898D
(Paper)
RSC Adv., 2012,
2, 12915-12921
C-HETSERF: distinction of cis/trans-isomers and measurement of long range couplings between chemically equivalent nuclei in polycyclic aromatic hydrocarbons†
Received
22nd August 2012
, Accepted 24th October 2012
First published on 25th October 2012
Abstract
The scalar couplings between chemically equivalent protons in symmetric molecules are not reflected in the NMR spectrum. The present study reports the utility of our previously reported two dimensional spin selective correlation experiment to measure the short and long range homo- and hetero-nuclear scalar couplings in such systems. In weakly coupled spin systems, the experiment also yields the relative signs of the couplings. The relative configurations of double bonded symmetrically disubstituted isomers could be determined from the measured long range vicinal proton-proton couplings (3JHH), owing to the fact that these couplings are consistently larger for trans isomers. The study also reveals that long range 2JCH cannot be utilized as an exclusive parameter for the identification of symmetrically disubstituted cis/trans isomers. The application of the methodology for the determination of couplings between the chemically equivalent protons in polycyclic aromatic hydrocarbons and in bigger molecules such as porphyrin, has also been demonstrated. The study, carried out on large number of molecules, shows the generality and wide applicability of the C-HETSERF experiment.
Introduction
There has been a growing importance in the study of naturally occurring C2 symmetric molecules and polycyclic aromatic hydrocarbons (PAHs) owing to their environmental and biological activities.1 Consequently, the identification of individual components from a mixture of symmetric molecules and their separation are of profound importance. Furthermore, the presence of two or more equivalent sub-units in symmetric molecules often creates very difficult analytical problems. Among the various analytical techniques generally employed in determining the structures of such molecules, NMR spectroscopy has played a dominant role.2 A serious limitation in the analysis of NMR spectra of such molecules is the detection of very few transitions due to chemical equivalence, hindering their assignment and subsequent structure elucidation. On the other hand, the vicinal proton–proton scalar coupling (3JHH) has been demonstrated to be an important spectral parameter for the distinction of cis- and trans-isomers as, for example, in symmetrically disubstituted alkenes. However, the inherent problem is the scalar couplings between chemically equivalent protons in such symmetric systems, as although they exist, they are not reflected in the conventional one dimensional (1D) 1H NMR spectrum. In favourable situations, they could be extracted from the well-resolved and non-overlapped 13C satellites of the 1D proton spectrum.3 In bigger molecules, the identification of satellites and the extraction of this information is formidable since, many a times, the satellite peaks are masked by the intense signals from the 12C bound protons. The precise assignment of the cis/trans-isomers in symmetrically disubstituted ethylenes, (R′RC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CRR′), employing the nuclear Overhauser effect (nOe) experiment, though feasible, is also a challenging task. One may encounter situations where there is overcrowding of the spectral lines or peaks, which is due to the absence of coupling fine structures in the NOESY spectra, restricting their analyses.4
CRR′), employing the nuclear Overhauser effect (nOe) experiment, though feasible, is also a challenging task. One may encounter situations where there is overcrowding of the spectral lines or peaks, which is due to the absence of coupling fine structures in the NOESY spectra, restricting their analyses.4
Although a plethora of 2D experiments have been reported for the study of symmetric molecules, they do not facilitate the measurement of long-range heteronuclear scalar couplings (nJXH).5–9 On the other hand, nJXH (n = 2, 3) has emerged as reliable parameters in the assignment of relative configurations and conformations.10 Earlier their utilization was limited due to the non-availability of appropriate NMR experimental techniques to measure such couplings, which are usually small, with the required accuracy. The recent developments of several two dimensional experimental methodologies have drastically improved this scenario.10d In the present study, we have applied the sign-sensitive C-HETSERF experiment to derive both short and long range homo- and hetero-nuclear scalar couplings (nJXH, where X = 13C/1H, n > 1).11–13 Subsequently the 3JHH and nJCH (n > 1) obtained from the 2D C-HETSERF spectrum are employed for the assignment of cis/trans-isomers. In addition, the utility of the method has been demonstrated by the measurement of short and long-range J-couplings among the chemically equivalent nuclei in bigger molecules, such as porphyrin and PAHs.
Experimental section
The experiments have been carried out on three mixtures of isomers, viz., cis and trans-stilbene (1 and 2, respectively), fumaric acid (3) and maleic acid (4), cis and trans-dichloroethene (5 and 6, respectively) and other symmetric molecules, such as, but-2-yne-1,4-diyl bis(perfluorophenyl) dicarbonate (7), phenanthrene (8), pyrene (9) and porphyrin (10). The chemical structures of the investigated molecules are reported in Scheme 1. The one and two dimensional spectra were recorded either on a Bruker DRX 500 MHz or AVANCE-400 MHz NMR spectrometer equipped with a TXI probe. The temperature was regulated at 298 K by using a BVT 3000 temperature control unit. The C-HETSERF pulse sequence,11–13 given in Fig. 1, was employed to obtain both homo- and hetero-nuclear couplings. A diagrammatic illustration of the information derivable10,13 from the diagonal and cross peaks of a weakly coupled three spin system of the type AMX is also given in Fig. 1.
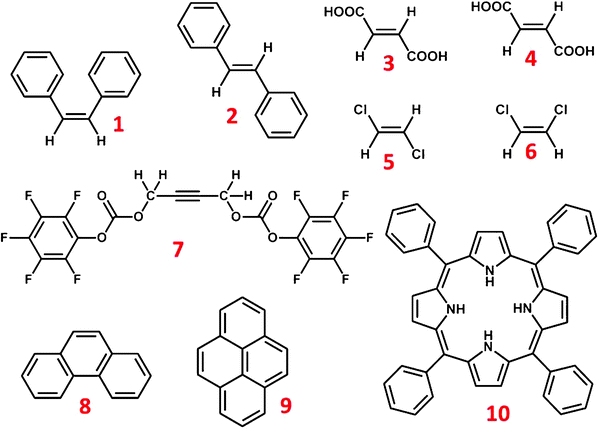 |
| | Scheme 1 Chemical structures of investigated molecules, cis-stilbene (1), trans-stilbene (2), fumaric acid (3), maleic acid (4), cis-dichloroethene (5), trans-dichloroethene (6), but-2-yne-1,4-diyl bis(perfluorophenyl) dicarbonate (7), phenanthrene (8), pyrene (9) and porphyrin (10). | |
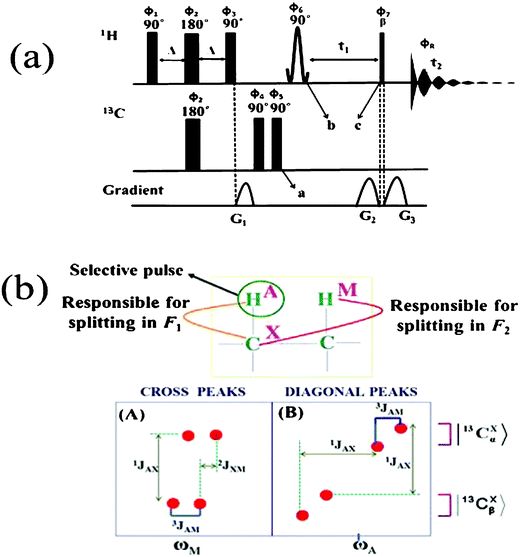 |
| | Fig. 1 (a) Pulse sequence utilized for the C-HETSERF experiment. The delay Δ responsible for the polarization transfer that depends on the factor 1/(4 × 1JCH). (b) Pictorial depiction of the extraction of different couplings from the C-HETSERF experiment in an AMX spin system when the selective pulse is applied on A spin; (A) The diagonal peaks of the 2D spectrum yielding the one bond heteronuclear coupling (1JAX). It is the frequency difference between the 13C-bound proton signals at their |13Cα> or |13Cβ> spin states in both the dimensions, (B) The cross peaks yielding the long range heteronuclear coupling (2JXM). The F2 cross-section of the 13C-attached proton signals at either |13Cα> or |13Cβ> spin states provides the homonuclear coupling between A and M (3JAM). In the all present experiments, a non-selective 90° pulse is applied on protons instead of a selective pulse before the t1 evolution. | |
In summary, it is a correlation experiment that connects 13C edited proton magnetization in both the dimensions. The anti-phase proton magnetization (2IxSz) is allowed to evolve under both short and long range proton–proton and proton–carbon couplings in both dimensions, facilitating their determination. The diagonal peaks yield one bond proton–carbon couplings and homonuclear couplings, whereas the cross peaks provide long range proton–carbon couplings in addition to proton–proton couplings. Since the pattern of the cross peaks is of the E-COSY type,14,15 the methodology also provides information on the relative signs of the couplings in weakly coupled spin systems from the slopes of the displacement vectors of the cross peaks. As far as the strongly coupled spin systems are concerned, the question of determination of relative signs of the couplings does not arise. A detailed description of the spin dynamics during the pulse sequence is provided in the ESI.† The delay “Δ” responsible for creation of the 13C-bound proton signal, other experimental and data processing parameters for all the experiments is tabulated in the ESI.†
Results and discussion
Assignment of symmetrically disubstituted cis-, trans-isomers
In symmetric molecules viz., Ha–12C–12C–Hb, containing two chemically equivalent protons, Ha and Hb, the chemical shift difference between Ha and Hb is zero and therefore individual chemical shifts are unobservable in the NMR spectrum. Although coupling between Ha and Hb exists, it does not get reflected in the spectrum, thereby hindering its measurement. Since JHaHb is much larger than ΔδHaHb (= 0), it constitutes a strongly coupled AA′ two spin system. On the other hand, if Ha is directly bonded to a 13C, spin (X), the spin symmetry is broken and can be treated as an ABX system. The transformation of the AA′ spin system to ABX is schematically illustrated for a symmetrically disubstituted ethylene molecule in Fig. 2.
 |
| | Fig. 2 The schematic illustration of coupled proton two spin system of the type AA′ getting transformed into the ABX type with the inclusion of one naturally abundant 13C spin. | |
Though the coupled protons and the 13C (i.e., Ha–13C–12C–Hb) form a spin system of the type ABX, even in this situation the chemical shift difference between the two protons arising due to isotopic effects by the inclusion of naturally abundant 13C is negligibly small [ΔδHaHb (= 0)] and is unobservable. Therefore, a single peak is detected even for this spin system in the 1H spectrum. On the other hand, the low intensity 13C satellite signals of Ha (Ha1 and Ha2) appear on either side of the Ha peak (attached to 12C), and the peak Ha1 gets separated from the Hb chemical shift by the distance 1JCH/2. As a consequence of coupling between Ha and Hb, the peak Ha1 splits into a doublet, enabling the direct measurement of JHaHb.
For the purpose of demonstrating the utility of C-HETSERF experimental methodology both for the determination of short and long range couplings and their utility for the assignment of relative configurations of double bonded symmetrically disubstituted molecules, a prepared 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of cis-stilbene (1) and trans-stilbene (2) was chosen. Non-aromatic protons of each of these molecules pertain to an AA′ spin system. The well-resolved 13C satellites, marked with red arrows, shown for the trans-isomer in Fig. 3, provide 3JHH. The measurement of 3JHH for the cis-isomer by identification of satellites corresponding to this molecule facilitates the distinction between the cis- and trans-isomers, since it is well established that (3JHH)trans > (3JHH)cis. The measurement of long-range 2JCH is, however, not possible from this spectrum as they are masked by the proton signals arising from the molecules attached to the abundant 12C spin. Consequently, for the simultaneous measurement of 3JHH and 2JCH, and their subsequent utilization for unambiguously distinguishing the isomers, the C-HETSERF experiment has been implemented. In C-HETSERF, the natural abundant 13C is introduced as a spy nucleus in the chemically equivalent proton spin system.12 This results in the breaking down of the symmetry of the AA′ spin system, and the three spin system then pertains to an ABX type, as schematically depicted in Fig. 2, where A and B are chemically non-equivalent protons and X is the 13C nucleus directly bonded to proton A. The scalar couplings measurable from the cross-peaks of the C-HETSERF spectrum reported in Fig. 4 are; 1JCH, 2JCH and 3JHH. The frequency difference between the displaced α/β-cross peaks along the direct dimension (marked as ‘a’ and ‘b’ for molecules 2 and 1, respectively) provides 2JCH. The frequency difference between two adjacent cross peaks in the same cross-section provides 3JHH (marked as ‘c’ and ‘d’ for 2 and 1, respectively). The separation ‘c’ and ‘d’ measured from the spectrum are 16.2 Hz and 12.2 Hz, respectively. The spectrum with a larger coupling (3JHH = 16.2 Hz) is assigned to the trans-isomer, unambiguously distinguishing it from its cis-counterpart. Therefore, 2JCH, pertaining to separations ‘b’ (2.7 Hz) and ‘a’ (3.8 Hz) could be assigned to the cis- and trans-isomers respectively.
:1 mixture of cis-stilbene (1) and trans-stilbene (2) was chosen. Non-aromatic protons of each of these molecules pertain to an AA′ spin system. The well-resolved 13C satellites, marked with red arrows, shown for the trans-isomer in Fig. 3, provide 3JHH. The measurement of 3JHH for the cis-isomer by identification of satellites corresponding to this molecule facilitates the distinction between the cis- and trans-isomers, since it is well established that (3JHH)trans > (3JHH)cis. The measurement of long-range 2JCH is, however, not possible from this spectrum as they are masked by the proton signals arising from the molecules attached to the abundant 12C spin. Consequently, for the simultaneous measurement of 3JHH and 2JCH, and their subsequent utilization for unambiguously distinguishing the isomers, the C-HETSERF experiment has been implemented. In C-HETSERF, the natural abundant 13C is introduced as a spy nucleus in the chemically equivalent proton spin system.12 This results in the breaking down of the symmetry of the AA′ spin system, and the three spin system then pertains to an ABX type, as schematically depicted in Fig. 2, where A and B are chemically non-equivalent protons and X is the 13C nucleus directly bonded to proton A. The scalar couplings measurable from the cross-peaks of the C-HETSERF spectrum reported in Fig. 4 are; 1JCH, 2JCH and 3JHH. The frequency difference between the displaced α/β-cross peaks along the direct dimension (marked as ‘a’ and ‘b’ for molecules 2 and 1, respectively) provides 2JCH. The frequency difference between two adjacent cross peaks in the same cross-section provides 3JHH (marked as ‘c’ and ‘d’ for 2 and 1, respectively). The separation ‘c’ and ‘d’ measured from the spectrum are 16.2 Hz and 12.2 Hz, respectively. The spectrum with a larger coupling (3JHH = 16.2 Hz) is assigned to the trans-isomer, unambiguously distinguishing it from its cis-counterpart. Therefore, 2JCH, pertaining to separations ‘b’ (2.7 Hz) and ‘a’ (3.8 Hz) could be assigned to the cis- and trans-isomers respectively.
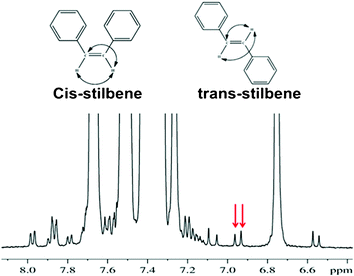 |
| | Fig. 3 500 MHz 1H spectrum of the mixture of cis- and trans-stilbene in the solvent CDCl3. The two 13C satellites for the trans-isomer are marked by red arrows. It may be noted that many satellites are hidden below the strong peaks from the 12C protons, severely hampering their identification. | |
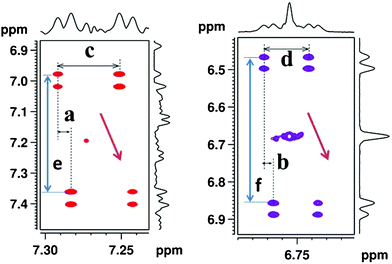 |
| | Fig. 4 400 MHz 2D C-HETSERF spectrum corresponding to the prepared mixture of samples 1 and 2. Selected cross-peaks regions are shown. The separations ‘e’ and ‘f’, along the indirect dimension, provide 1JCH, whereas separations ‘a’ and ‘b’ (displacement of cross peaks), along the direct dimension, provide 2JCH. Since the relative slopes of the displacement vectors are identical (as marked by brown arrows) the relative signs of the 2JCH values are the same in both molecules 1 and 2. | |
The relative slopes of the displacement vectors of the cross peaks (indicated by the tilted brown coloured arrows in Fig. 4) along the direct dimension indicate that the relative signs of 2JCH, in both the molecules 1 and 2, are identical. For demonstrating the wider utility of this experiment, investigations were carried out on other mixtures of symmetrically disubstituted molecules, such as 3 and 4, and 5 and 6. The homo- and hetero-nuclear couplings determined for these molecules are reported in Table 1.
Table 1 Determined scalar coupling constants (3JHH and 2JCH in Hz) for the investigated molecules. The accuracy of determination of 3JHH and 2JCH are given by LW/SN, where LW is the line width and SN the signal-to-noise ratio.16 For molecules 8 and 9, it may be pointed out that J values are very much less than the digital resolution. These values are not measured from a single cross section. They are measured from the displacement of cross sections arising due to spin state selection and are accurate
Can 2JCH be an indicator for the assignment of cis- and trans-isomers?
As a consequence of the determination of long range heteronuclear couplings (2JCH) by C-HETSERF, an intriguing question that remains to be answered is, whether this parameter can also be employed as an indicator for the assignment of cis- and trans-isomers, either exclusively or in conjunction with 3JHH. Hence a systematic study was carried out on the pairs of symmetrically disubstituted cis- and trans-isomers. From the derived parameters reported in Table 1, it was observed that in fumaric (3) and maleic acid (4), similar to cis/trans-stilbene (1/2), the 2JCH for the trans-isomer (3) is consistently larger than its cis-counterpart (4). On the other hand for cis- and trans-dichloroethene (5 and 6, respectively), the situation gets reversed. In molecules 5 and 6, the 2JCH is larger for the cis-isomer than the trans-isomer. Therefore, it evidently establishes the fact that unlike 3JHH, the 2JCH can neither be construed as an exclusive parameter nor be utilized in conjunction with 3JHH for identification of cis/trans-isomers.
Application of C-HETSERF to symmetric molecules
The approach reported in this work is not only confined to the study of cis/trans-isomers but is also applicable to other types of symmetric isomers possessing methyl, methylene and methine groups. As an example, the analysis of NMR spectra of a symmetric molecule, such as but-2-yne-1,4-diyl bis(perfluorophenyl) dicarbonate (structure is given in Scheme 1), using routinely employed techniques, such as HSQC, HMQC, etc., is very difficult.3 Furthermore, the nOe experiment cannot be applied as this is not detectable between the homotopic methylene protons. On the other hand, the analysis of the spectrum and subsequent extraction of the couplings is possible using the C-HETSERF experiment (the C-HETSERF spectrum is given in the ESI†). The 1H-NMR signals of but-2-yne-1,4-diyl bis(perfluorophenyl) dicarbonate comprises of a triplet due to the five bond long-range coupling (5JHH) with two other protons. The 5JHH thus determined is 1.8 Hz. It is very interesting to note that in this symmetric molecule, we could precisely determine the 4JCH of 1.2 Hz, which otherwise would have been impossible from the conventional 1H spectrum.
Application to polycyclic aromatic hydrocarbons (PAHs) and porphyrin
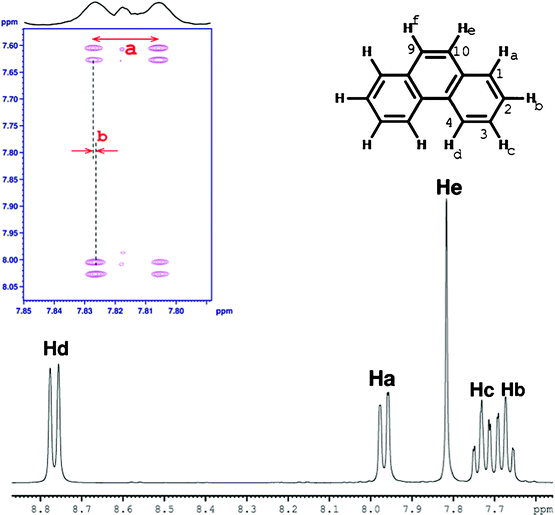
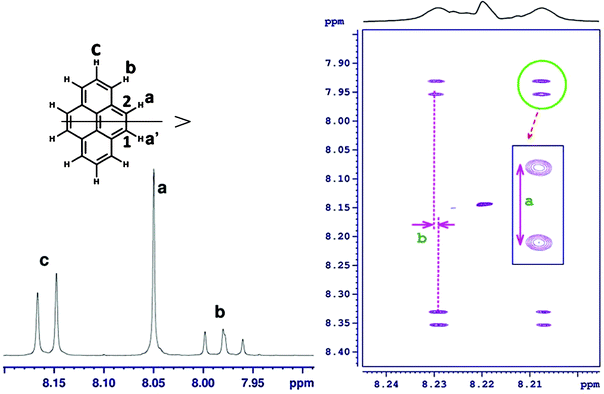
Another interesting application of C-HETSERF is in the determination of couplings between identical segments of polycyclic aromatic hydrocarbons. As an example, the protons marked He and Hf in the phenanthrene molecule (8) (Fig. 5) are equivalent and appear as a singlet at 7.81 ppm, rendering it possible to determine the coupling between them. However, the C-HETSERF spectrum reported in Fig. 5 exhibits the coupling between them. The separations giving couplings 3JHeHf and 2JC10Hf are marked as ‘a’ and ‘b’ in the spectrum and their measured values are 8.6 Hz and 0.2 Hz, respectively. This brings out another immediate advantage of the C-HETSERF experiment, wherein the coupling of such a small magnitude could be measured accurately as they are measured from the displacement of cross peaks in the direct dimension (they are separated by 1JCH in the indirect dimension).
An additional example is the molecule pyrene (9). In this system the coupling between the symmetric protons (marked Ha and Ha′ given in Fig. 6) cannot be determined by conventional NMR experiments. However, the C-HETSERF cross peaks at Ha, reported in Fig. 6, provides the coupling between them. The measured couplings 3JHaHa′ and 2JC1Ha (marked as ‘a’ and ‘b’ in Fig. 6) are 8.7 Hz and 0.2 Hz, respectively. In an identical manner, the method can be extended to measure the couplings between the symmetric units in other reasonably bigger PAHs. Another commonly encountered problem is that other methods, such as COSY and NOESY, are insufficient to assign all the chemical shifts in the 1H and 13C NMR spectrum of PAHs.9a However, the long range correlations (nJCH) could be determined from this experiment and utilized to assign closely resonating 13C chemical shifts.17
 |
| | Fig. 6 (Left) 400 MHz 1H spectrum of pyrene in the solvent CDCl3. (Right) The region of the C-HETSERF spectrum corresponding to the chemical shift of the proton marked Ha. | |
Another advantage of this methodology is exploited in the study of porphyrin molecules, where the coupling between the symmetric protons of the pyrrole moiety cannot be measured from the conventional one dimensional spectrum. The cross peak of the C-HETSERF spectrum pertaining to these protons is reported in Fig. 7. The measured couplings 3JHH and 2JCH (marked as ‘a’ and ‘b’ in Fig. 7) are 4.1 Hz and 4.0 Hz, respectively.
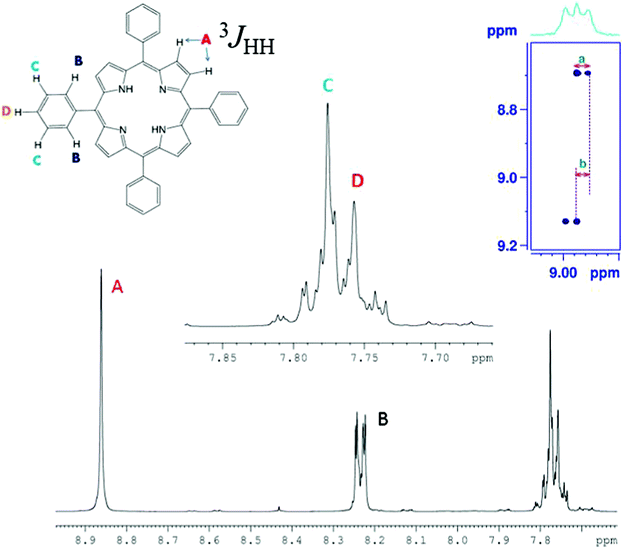 |
| | Fig. 7 400 MHz 1H spectrum of porphyrin in the solvent CDCl3. Assignments of chemical shifts to different protons are marked with letters. The region of the C-HETSERF spectrum corresponding to chemical shift of proton marked Ha. | |
Conclusions
The utility of the C-HETSERF experiment in the measurement of long-range proton–proton and carbon–proton scalar couplings and also in achieving the distinction between symmetrically disubstituted double bonded isomers is demonstrated on the basis of 3JHH. However, unlike the well known concept of (3JHH)trans > (3JHH)cis, an anomalous pattern is encountered in the 2JCH couplings, that are not consistently large for one type of isomer, establishing that 2JCH cannot be construed as an exclusive parameter or be used in conjunction with 3JHH for the assignment of symmetrically disubstituted cis/trans-isomers. The couplings of smaller magnitudes, that are generally hidden within the line widths in the one dimensional proton spectrum, could be accurately determined from the displacement of α/β cross peaks in the direct dimension. In weakly coupled spin systems, the direction of displacement of the cross sections can be utilized to obtain the relative signs of the couplings. The wider utility of the C-HETSERF experiment in the determination of couplings between the chemically equivalent protons in larger polycyclic aromatic hydrocarbons, and in porphyrin, has also been demonstrated.
Acknowledgements
SRC would like to thank the UGC for the SRF, and NN thanks the IISc for a research associateship. NN thanks Dr Suresh Kumar Vasa for useful discussions. NS gratefully acknowledges the generous financial support of the Science and Engineering Research Board, New Delhi (grant no. SR/S1/PC-42/2011).
References
-
(a) G. Balanikas, N. Hussain, S. Amin and S. S. Hecht, J. Org. Chem., 1988, 53, 1007 CrossRef CAS;
(b) R. E. Lehr, P. L. Kole, M. Singh and K. D. Tschappat, J. Org. Chem., 1989, 54, 850 CrossRef CAS;
(c) A. S. Kiselyov, H. Lee and R. G. Harvey, J. Org. Chem., 1995, 60, 6123 CrossRef CAS.
-
(a) F. D. Gunstone, M. R. Pollard, C. M. Scrimgeour and H. S. Vedanayagam, Chem. Phys. Lipids, 1977, 18, 115 CrossRef CAS;
(b) R. Rossi, A. Carpita, M. G. Quirici and C. A. Verancini, Tetrahedron, 1982, 38, 639 CrossRef CAS;
(c) N. M. Carballeira and F. Shalabi, Lipids, 1990, 25, 835 CrossRef CAS;
(d) N. M. Carballeira and C. Cruz, Chem. Phys. Lipids, 1996, 84, 81 CrossRef CAS;
(e) H. Heckers, F. W. Melcher and U. Schloeder, J. Chromatogr., A, 1977, 136, 311 CrossRef CAS;
(f) H. Jaeger, H. U. Klor and H. J. Ditschuneit, J. Lipid Res., 1976, 17, 185 CAS.
-
(a)
H. Günther, NMR Spectroscopy: Basic Principles, Concepts, and Applications in Chemistry; John Wiley and Sons, New York, 1995, p. 214 Search PubMed;
(b) V. S. Fluxa, T. A. Jenny and C. G. Bochet, Tetrahedron Lett., 2005, 46, 3793 CrossRef CAS.
- L. Lunazzi and A. Mazzanti, J. Am. Chem. Soc., 2004, 126, 12155 CrossRef CAS.
-
(a) R. T. Williamson, J. R. Carney and W. H. Gerwick, J. Nat. Prod., 2000, 63, 876 CrossRef CAS;
(b) R. C. Crouch, R. B. McFayden, S. M. Daluge and G. E. Martin, Magn. Reson. Chem., 1990, 28, 792 CrossRef CAS;
(c) D. Yang, Y. Daiwen, X. Xu and C. Ye, Magn. Reson. Chem., 1993, 31, 585 CrossRef;
(d) S. R. Salman, R. D. Farrant, P. N. Sanderson and J. C. Lindon, Magn. Reson. Chem., 1992, 32, 585 Search PubMed;
(e) S. Simova, Magn. Reson. Chem., 1998, 36, 505 CrossRef CAS.
- A. Poveda, C. Vicent, S. Penades and J. J. Barbero, Carbohydr. Res., 1997, 301, 5 CrossRef CAS.
-
(a) J. Kawabata and E. J. M. Fukushi, J. Am. Chem. Soc., 1992, 114, 1115 CrossRef CAS;
(b) R. Wagner and S. Berger, Magn. Reson. Chem., 1997, 35, 199 CrossRef CAS;
(c) A. Rajca and M. Rabinovitz, Magn. Reson. Chem., 2000, 38, 311 CrossRef;
(d) R. M. Gschwind, X. Xie and P. R. Rajamohan, Magn. Reson. Chem., 2004, 42, 308 CrossRef CAS and reference therein.
- P. Nolis, A. Roglans and T. Parella, Magn. Reson. Chem., 2005, 43, 979 CrossRef CAS.
-
(a) R. E. Hoffman, R. Shenhar, I. Willner, H. E. Bronstein, L. T. Scott, A. Rajca and M. Rabinovitz, Magn. Reson. Chem., 2000, 38, 311 CrossRef CAS;
(b) R. Wagner and S. Berger, Magn. Reson. Chem., 1997, 35, 199 CrossRef CAS.
-
(a) M. Eberstadt, G. Gemmecker, D. F. Mierke and H. Kessler, Angew. Chem., Int. Ed. Engl., 1995, 34, 1671 CrossRef CAS;
(b) N. Matsumori, D. Kaneno, M. Murata, H. Nakamura and K. Tachibana, J. Org.
Chem., 1999, 64, 866 CAS;
(c) P. Vidal, N. Esturau, T. Parella and J. F. Espinosa, J. Org. Chem., 2007, 72, 3166 CrossRef CAS and references therein;
(d) N. Nath and N. Suryaprakash, ChemPhysChem, 2012, 13, 645 CrossRef CAS.
- N. Nath and N. Suryaprakash, Chem. Phys. Lett., 2011, 502, 136 CrossRef CAS.
- N. Nath and N. Suryaprakash, J. Phys. Chem. B, 2011, 115, 6868 CrossRef CAS.
-
N. Nath, PhD thesis, Indian Institute of Science, Bangalore, 2011.
- C. Griesinger, O. W. Sørensen and R. R. Ernst, J. Magn. Reson., 1987, 75, 474 CAS.
- C. Griesinger, O. W. Sørensen and R. R. Ernst, J. Magn. Reson., 1986, 64, 6873 Search PubMed.
- P. Trigo-Mourino, A. Navarro-Vazquez, J. Ying, R. R. Gil and A. Bax, Angew. Chem., Int. Ed., 2011, 50, 7576 CrossRef CAS.
- A. Bax, J. Magn. Reson., 1984, 57, 314 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Description of C-HETSERF experiment, experimental and processing parameters and related references. See DOI: 10.1039/c2ra21898d |
| ‡ Present Address: Dept. of NMR-based Structural Biology, Max-Plank Institute for Biophysical chemistry, Göttingen, Germany. |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.