Soft coordination supramolecular polymers: novel materials for dual electro-catalysis†
Yawei
Liang‡
a,
Jing
Zhao‡
ab,
Dewen
Zhang
a,
Yun
Yan
*a,
Yinglin
Zhou
*a,
Xinxiang
Zhang
a and
Jianbin
Huang
*a
aBeijing National Laboratory for Molecular Sciences (BNLMS), Key Laboratory of Biochemistry and Molecular Engineering, College of Chemistry and Molecular Engineering, Peking University, Beijing, 100871, P. R China. E-mail: yunyan@pku.edu.cn; zhouyl@pku.edu.cn; jbhuang@pku.edu.cn
bDepartment of Chemistry, University of California, One Shields Avenue, Davis, CA 95616, USA
First published on 3rd October 2012
Abstract
We report in this paper a dual catalytic effect of water soluble soft coordination supramolecular polymers (SCSPs) based on Fe3+ and a bis-ligand. Upon assembling the Fe(III)-SCSPs onto electrodes via layer-by-layer technique, the electroactive films may lower the half-wave potential for all the tested molecules and enhance the current for neutral or oppositely charged electroactive species. The decrease of the half-wave potential was attributed to the mediation of the Fe(III)-SCSPs in electron transferring, whereas the enhancement of current is a consequence of accumulation of electrons in the films. We verified that this dual catalytic effect is general to neutral and cationic redox active molecules, and can be used to detect hydrogen peroxide owing to the significant enlargement of catalytic current. Our results proved that the water soluble soft coordination supramolecular polymers containing metal centers are indeed a novel class of advanced catalytic materials, which may open a new vista on the design and study of materials of this class.
Introduction
Supramolecular polymeric structures containing metal centers are emerging as an interesting and broad class of easily processable material with properties and functions that enhance those of pure organic ones.1 One can find nanoscale coordination polymer particles2–5 as well as linear or three-dimensional soluble structures that are self-assembled from metal ions and polytopic organic ligands,6–11 which we would like to call as soft coordination supramolecular polymers (SCSPs). Recent studies indicate that SCSPs may be used as self-healing materials owing to the dynamic nature of coordinating bonds.8,11 In the past decade, various SCSPs of transition and rare earth metals with judicious ligands have been achieved.12,13 It is expected that these metal-containing novel soft materials may exhibit a number of attractive properties such as luminescence,14–18magnetic cross-over,19redox responsiveness,20–22 and catalytic performance.23,24 So far, studies have verified that SCSPs indeed exhibit interesting light emitting properties which take advantage of both the metal ions and the ligands.9,15,17,18 It was found that upon doping various metal ions in one polymeric network, the photo luminescent behaviors can be finely tuned.16 More interestingly, upon delicate design of ligand structures, fluorescent materials are able to be produced when Fe2+, which is an efficient quencher to most fluorescence, was used to build SCSPs with large organic ditopic ligands.25 Meanwhile, studies in our group suggest that the molecular assemblies based on iron containing SCSPs may exhibit redox responsiveness.20–22The aforementioned work unambiguously proved that some of the expectations on SCSPs have been made into reality. However, it is very surprising that so far no valuable catalytic properties of these materials have been reported. The only attempt made so far was based on a so-called Co-MEPE, which was assembled from Co2+ and a rigid ditopic ligand 1, 4-bis (2,2′:6′,2′′-terpyridine-4′-yl) benzene.26 However, it was found that this modification layer of Co-MEPE simply blocks the channel for efficient communication of electrons between Fe(CN)63−/4− and the electrode without showing an electrocatalytic effect. Since the terpyridine ligand is hydrophobic, the polarity inside the film was lowered considerably, which was confirmed by the fluorescence spectra of pyrene.26 This is probably the reason why electroactive molecules were blocked by the films. It is therefore reasonable to expect an electrocatalytic effect from a water soluble ligand. Actually, Schlenoff et al. verified that the current for water electrolysis can be significantly enhanced if a gold electrode was modified by a covalent redox-active polyelectrolyte multilayer.27 These inspired us to study the electrocatalytic properties of the water soluble SCSPs developed in our group.
In this study we show that when a water soluble Fe(III)-SCSP (Scheme 1) was modified onto a glassy carbon electrode (GCE) or a fluorine doped tin oxide electrode (FTOE), one may observe either a decrease of the half-wave potential, or an enhancement of the catalytic current. This effect can be observed for both positively charged and charge neutral probe molecules. Our results suggest that water soluble redox-active SCSPs show dual catalytic effect, which may open up a new vista for new electrocatalytic materials.
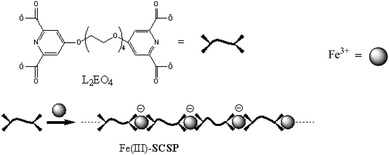 | ||
| Scheme 1 Formation of soft coordination suprapolymer Fe(III)-L2EO4 from Fe3+ and ditopic ligand L2EO4 in water. | ||
2 Experimental
2.1 Materials
The water soluble Fe(III)-SCSPs were prepared simply by mixing the aqueous solution of bis-ligand L2EO4 (Scheme 1) and fresh FeCl3 solution at equimolar ratio as described in our previous work.28,29 K4Fe (CN)6, dopamine, and KNO3 were purchased from Sigma-Aldrich. Polyethylenimine (PEI, branched), and polystyrene sulphite, sodium salt, (PSS), were purchased from Aldrich with Mw of 25,000. Milli-Q water was used throughout the work.2.2 Electrode modification
For the modification of electrodes used in this study, alternatively dropping 10 μL aqueous solution of PEI and that of Fe(III)-L2EO4 on a glassy carbon electrode (GCE) or a fluorine doped tin oxide (FTO) glass slide was adopted. Both solutions contain 0.1 M KNO3, which is the supporting electrolyte for electrochemical experiments. Prior to assembly, GCE was first polished with 0.05 mm alumina slurry by a polisher and then washed in H2O and ethanol for 3 mins in an ultrasonic cleaner. FTO glass slide was sonicated in the mixed solvent of Lysol and Milli-Q water (volume ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3), then sonicated 3 times in Milli-Q water each for another 30 mins before uses. PEI was deposited as the first layer on both the GCE and FTO slide. Then the surface was washed with Milli-Q water three times each for 1 min and dried by argon. Next, the alternative assembly of Fe(III)-L2EO4 and PEI was carried out. The concentration of Fe(III)-L2EO4 and PEI is 2 mM and 10 mM, and the assembling time is 15 and 1 min, respectively.
:3), then sonicated 3 times in Milli-Q water each for another 30 mins before uses. PEI was deposited as the first layer on both the GCE and FTO slide. Then the surface was washed with Milli-Q water three times each for 1 min and dried by argon. Next, the alternative assembly of Fe(III)-L2EO4 and PEI was carried out. The concentration of Fe(III)-L2EO4 and PEI is 2 mM and 10 mM, and the assembling time is 15 and 1 min, respectively.
2.3 Electrochemical measurements
A CHI660C electrochemical workstation (Shanghai Chenhua Equipments, China) with a conventional three-electrode system was used to perform electrochemical measurements. A platinum wire was used as a counter electrode, and Ag/AgCl as a reference electrode. The scan rate for CV analysis was 100 mV s−1 except for the study of scan rate dependence. Coated GCE (0.07 cm2) was used as the working electrode for measuring the CV behaviors of the electroactive probes, whereas a coated FTO slide (10 mm × 30 mm) was employed for UV measurements and electrochemical tests for methyl viologen (MV2+) ions.Voltammetric and impedance data were collected for GCE after each assembling step. The (modified) electrode was rinsed with pure water before assembling next layer. Charge transfer resistance (Rct) values were obtained by fitting impedance data with a Randles circuit which included the electrolyte resistance between working and reference electrodes (Rs), the Warburg impedance (Zw), a constant phase element (Q) representing the double layer capacitance for the (modified) electrodes and the electron-transfer resistance (Ret).
2.4 UV/Vis measurement
Layer growth and changes stimulated by electrochemical redox stimuli on FTO slide were tracked by using a Shimadzu UV-2550 UV-spectrometer. Spectra in the range 330–500 nm were collected with a scanning rate of 10 nm min−1.3 Results and discussion
The coordination complexes of Fe3+ and L2EO4 carry one elementary negative charge at each coordination center,20,21 and are unable to form polymeric structures at low concentrations due to their concentration dependent degree of polymerization.13 However, our previous results demonstrate that this class of coordinating systems may automatically transform into polymeric structures in the presence of oppositely charged polyelectrolytes owing to the enhancement of local concentration.30,31 We have demonstrated that Fe(III)-L2EO4 can be successfully assembled onto quartz and silicon surfaces via layer by layer technique.32 In this study, the Fe(III)-L2EO4 coordination complexes were assembled with this strategy to GCE or FTO slide to study their electrochemical properties. The detailed characterization of the modified electrode can be found in Fig. S1–S3, ESI.†3.1 Electrochemical activity of Fe(III)-L2EO4 in layer-by-layer assemblies
First of all, the electrochemical characteristics of {PEI/Fe(III)-L2EO4}n films were monitored by cyclic voltammetry (CV). After each adsorption cycle creating a new PEI/Fe(III)-L2EO4 bilayer, the electrode was washed with pure water and then transferred to a 0.1 M KNO3 solution for CV measurements. A pair of well-defined, nearly reversible CV peaks is observed at about 0.035 V and 0.081 V, respectively, (Fig. 1A), which are characteristic of the Fe(III)/Fe(II) redox couples. The reduction and oxidation peak currents grow with the number of PEI/Fe(III)-L2EO4 layers (n) until n = 5. At n > 5, no further increase in the peak currents was observed, indicating that Fe(III)-L2EO4 in the bilayers above n > 5 is not addressed electrochemically. This is also an indication that the thickness of the films at n beyond 5 is too large to allow electron transportation. This is probably caused by the repulsion between the electrons and the coordination centers in the films. We have demonstrated in our previous work that upon reduction of the Fe3+ into Fe2+, the negative charge at each coordination center increases from −1 to −2. Therefore, the electrons may encounter increased repulsive forces with increasing numbers of assembled bilayers. It can be read from Fig. 1A that the separation between the anode and cathode potentials for {PEI/Fe(III)-L2EO4}5 films is about 0.041 V. This value is obviously smaller than that for a typical diffusion controlled one electron transfer process of 0.059 V, which suggests that electron hopping in the film is a thin-layer behavior.33 In addition, ipa and ipc increase linearly with scanning rate v, rather than with v0.5 (Fig. 1B),33 which also confirms a surface-confined thin-film behavior. However, the presence of electrostatic repulsive forces determines that the separation between the anode and the cathode potentials deviates considerably from zero, which is characteristic for typical thin-film electrochemistry.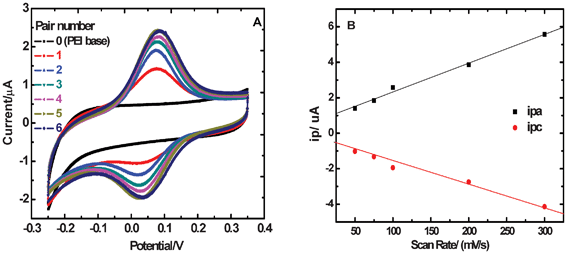 | ||
| Fig. 1 CV curves for {PEI/Fe(III)-L2EO4}n films on GCE. As n increases from 0 to 5, the peak current keeps increasing, whereas it does not increase at n ≥ 6 (A). Linear dependence of peak current on scanning rate (B). Original CV data at different scan rates is available in ESI.† | ||
To further check the electrochemical activity of Fe(III)-L2EO4 in the film, the current generated from electroactive Fe(III)-L2EO4 was measured when the film was exposed to a reduction (−0.25 V) and an oxidation (0.35 V) potential, alternatively. It can be read in Fig. 2A that as the potential varies, the current hops between negative and positive values without obvious decay up to several tens of cycles. In line with this, the characteristic UV absorbance of Fe(III)-L2EO4 at 380 nm decreases as the potential switches from −0.25 V to 0.35 V whereas the absorbance of Fe(II)-L2EO4 in the visible region increases (Fig. 2B). The combination of current cycle and UV absorbance suggests that electron transfer within the redox accessible layers in the film is reversible, and the film is very stable upon electrochemical stimulus. These allow further study of catalytic effect toward electroactive molecules.
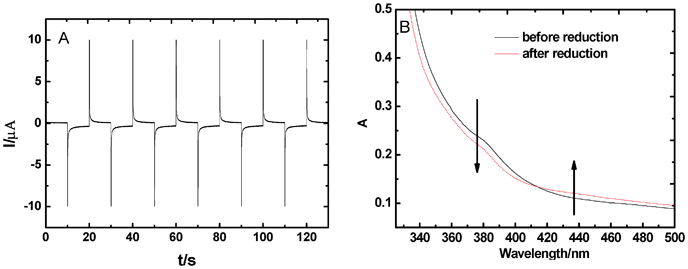 | ||
| Fig. 2 (A) Current of the {PEI/Fe(III)-L2EO4}6 films at FTO electrode during sequential dual-potential steps between −0.25 V and 0.35 V. (B) (b) UV-vis absorbance of the film corresponding to the two potentials: solid line, −0.25 V; dashed line: 0.35 V. | ||
3.2 Electrocatalytic study
Electrodes modified with redox active materials are expected to show a catalytic effect on electrochemical reactions. Increase of peak current for redox active molecules was observed very often on electrodes modified with polyoxometalates (POMs).20 To check the electrocatalytic behavior of Fe(III)-L2EO4, we chose two common electroactive probes. One is the negatively charged Fe(CN)64− ions, the other is the positively charged methyl viologen cations (MV2+).Firstly, CV plots for Fe (CN)64− at GCE were obtained. Surprisingly, as n of the {PEI/Fe(III)-L2EO4}n film increases from 1–5, one simply observes a decrease of the current of Fe(CN)63−/4− pair, which is accompanied by slightly increased peak separation (ΔEp) (Fig. 3A, curves for 0–4). Meanwhile, the CV curves become broad and plateau-shaped, suggesting current originating from slow diffusion of the Fe(CN)64− ions through the polyelectrolyte.35–37 it was noted that in these processes, the half-wave potential E1/2 keeps constant, indicating that the activation energy for this electrochemical reaction doesn't change. However, when the number of Fe(III)-L2EO4/PEI layer pairs ≥ 6, a considerable shift of the CV curve was observed, as demonstrated in Fig. 3A by the pink curve.
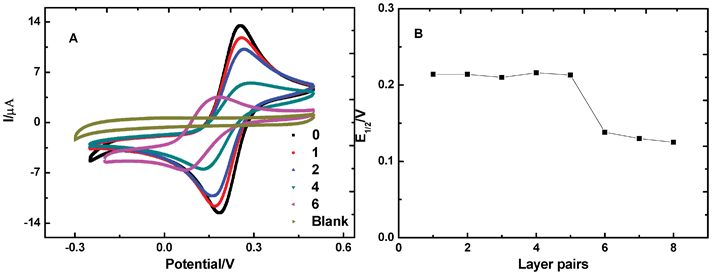 | ||
| Fig. 3 (A) CVs of 2 mM Fe (CN)64− solution on different GC electrodes. The numbers 0–6 demonstrate the number of {PEI/Fe(III)-L2EO4} layer pairs on the GC electrode; ‘blank’ represents the CV curve of {PEI/Fe(III)-L2EO4}6 films on the GC electrode with 0.1 M KNO3 as the supporting electrolyte. In all measurements, the scan rate is 0.1 Vs−1. (B) Variation of the half-wave potential with increasing numbers of {PEI/Fe(III)-L2EO4} layer pairs. | ||
On the surface of the unmodified GCE, the half-wave potential E1/2 of Fe(CN)63−/4− pair is 0.220 V; in contrast, on the 6-pair-modified electrode it becomes 0.138 V. The 0.220 V − 0.138 V = 82 mV decrease in E1/2 on the modified GCE corresponds to a decrease in Gibbs energy, which is 7.9 kJ mol−1 according to the following equation:
| ΔG = nΔEF |
However, it is noticeable that the current for the Fe(CN)63−/4− pair is rather low on the modified GCE, which is disadvantageous in catalytic reaction. We expect that such a low current is probably brought up by the repulsion between electrons and the co-charged Fe(CN)63−/4− pairs which decreased the efficiency of electron communication. To verify this point, the CV measurements for a positively charged redox active species, methyl viologen (MV2+), were performed. As can be seen in Fig. 4, the redox wave is a two-electron process on unmodified FTO slide, which corresponds to the following two reaction steps:

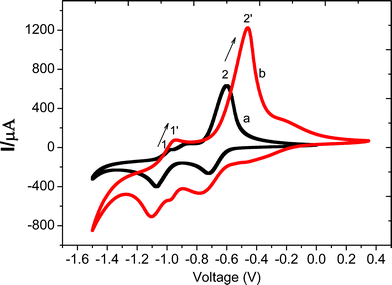 | ||
| Fig. 4 CV curves of MV2+ on (a) bare and (b) {PEI/Fe(III)-L2EO4}8 modified FTO electrode in 0.1 M KNO3 supporting electrolyte. | ||
As expected, a significant increase of the current was observed on the modified electrodes, which verified that the positively charged MV2+ may sequester the electrons more efficiently from the film. In addition, the half-wave potentials for the first and second electron transfer have been shifted toward zero by 15 and 45 mV, respectively, suggesting that the reduction of MV2+ becomes much easier. A similar increase of peak current was observed for the GC electrode modified with 6 layer pairs (see Fig. S4, ESI†) too, and the half-wave potentials for the two electron processes were shifted towards zero by 35 and 21 mV, respectively. Again, we found that no obvious shift of E1/2 occurs at the number of layer pairs below 5, and no increase of current for MV2+ was observed. These results indicate that the thickness of the films is very crucial for the occurrence of catalytic effect, which we will discuss in detail in the mechanism section. It should be noted that the shape of CV curves of MV2+ is markedly affected by the scan rate, the concentration, the nature of the electrode and the supporting electrolytes, so that it is common that their peak position, shape, and height appear differently in various scans.38 That is why we observed different peak numbers in the two curves in Fig. 4 and that in Fig. S4.†
Control experiments were made when the electroactive Fe(III)-L2EO4 was replaced by a non-electrochemical active covalent polyelectrolyte, PSS. As expected, we observed only a decrease of the peak current and a broadening of the CV curves with increasing layer numbers, but without any changes in half-wave potential (See Fig. S6, ESI†). This is characteristic of the resistance effect of permissive unconductive films, as reported by Bruening et al.35 This unambiguously supports our conclusion that electrochemical active Fe(III)-L2EO4 is crucial for catalytic phenomena to occur.
3.3 Mechanism of catalytic effect of Fe-L2EO4 modified electrode
The occurrence of a catalytic effect only at layer pairs beyond 6 indicates that the thickness of the film is very crucial, which may affect the depth where exchange of electrons occurs. According to literature,26,34 the thickness for a pair of oppositely charged polyelectrolyte in the presence of 500 mM salt is ∼5 nm; and we may estimate that the 5 pairs modified GCE has a thickness of about 25 nm. This is also in good agreement with our experimental results from ellipsommetry (data not shown). As demonstrated above, the electron exchange inside the modified electrode shows thin-film electrochemical characteristics, while the electrochemical behaviors of the Fe (CN)63−/4− pair and MV2+ on the modified electrode are controlled by diffusion processes. This suggests that the electroactive molecules have to diffuse into the films to encounter electrons migrating in thin-film mode. This means that acquiring electrons can occur in certain depths of the film away from the bare surface of the electrode. If the film is too thin, i.e., less than 25 nm or so in our study, there could still be uncovered sites on the electrode, so that the electroactive probes (here Fe(CN)63−/4− or MV2+) may diffuse quickly to the exposed electrode surface before the electron exchange occurs. Therefore, the redox reaction took place at the pristine GCE, and no catalytic behavior can be observed. In contrast, if the films are thick enough, there are no uncovered sites any more on the electrode. In this case, the diffusion of the probes becomes much slower so that the electrochemical processes occur at the surface of the films, rather than at the pristine GCE (Scheme 2). As a result, the coordination centers of Fe(III)-L2EO4 may serve as mediators in the redox process and facilitate the electrochemical reactions. | ||
| Scheme 2 Schematic representation of the electrochemical process occurred in the modified and unmodified GC electrodes. | ||
With regard to the increase of current for electroactive cations, it can be attributed to the accumulation of electrons in the reduction process. This can be confirmed by tracing the current with time under a constant reducing potential. We observed that the reduction current for both of the two electron-transfer processes of MV2+ becomes more negative with increasing reducing time; whereas such an effect cannot be observed if an oxidation potential was exerted (Fig. 5).
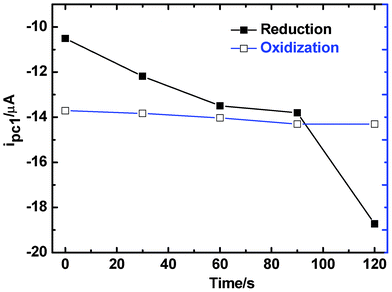 | ||
| Fig. 5 Variation of the peak current with time for the first electron transferring process of MV2+ on a {PEI/Fe(III)-L2EO4}6 modified GC electrode treated with a reducing (solid squares) and an oxidizing potential (empty squares), respectively. | ||
3.4 Generality of the dual catalytic effect and sensing physically important species.
The above catalytic mechanism indicates that the dual electrocatalytic effect should be observed for other electroactive species as long as the species do not carry negative charges. This was verified in the measurements of dopamine (DA), which is an important hormone neurotransmitter of redox activity. Due to its low electrochemical responsiveness, promotion of its redox current has attracted extensive studies from electrochemists.39 In Fig. 6 we show that the CV curve of DA gives two low and broad peaks when measured on a bare GCE. In contrast, on a GCE modified with 6 pairs of {PEI/Fe(III)-L2EO4}, the current was increased considerably and both the two peaks become very sharp. This suggests that the modified GCE electrode can be potentially used as a DA sensor. In addition, the oxidized potential for the first electron process was shifted 0.15 V towards the left, whereas the reduced one for the second electron process was moved 0.15 V towards the right. The changes in the CV curve of neutral DA, together with results for Fe(CN)63−/4− and MV2+, unambiguously demonstrate that Fe(III)-L2EO4 indeed exhibits a general dual catalytic effect to a large variety of electroactive species, which may find wide applications in related fields.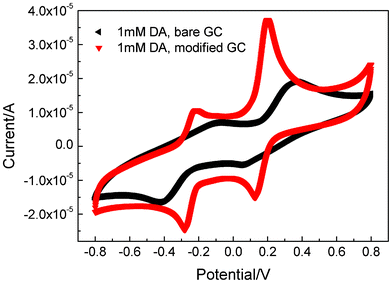
3.5 Application of the {PEI/Fe(III)-L2EO4} modified GCE in the detection of hydrogen peroxide
To further confirm this unique feature, the GCE modified with 6 pairs {PEI/Fe(III)-L2EO4} was applied to detect hydrogen peroxide since it plays an essential role as a mediator in many biological and environmental contamination processes.40 It is found that the current for the electrochemical reaction of hydrogen peroxide was promoted significantly (Fig. 7 A), whereas it was almost undetectable on the bare GCE. The Tafel curve (Fig. 7B) suggests that the current might be enhanced 10 times at an operating potential larger than 0.4 V. This means that under proper design, Fe(III)-L2EO4 may be exploited to detect the peroxide intermediates synchronized with many diseases.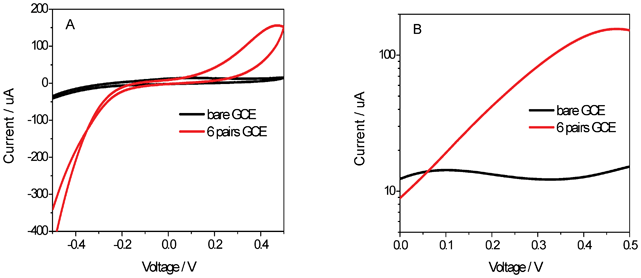 | ||
| Fig. 7 (A) CV curves of hydrogen peroxide solution with the modified GC. (B) Tafel Curves of modified GC and non-modified GC in Ar purged H2O2 solutions at room temperature. | ||
4 Conclusion
In summary, the water soluble supramolecular soft coordination polymer of Fe(III)-L2EO4 shows distinct electrocatalytic properties upon assembly onto electrodes via layer-by-layer technique. The presence of Fe(III)-L2EO4 inside the films decreases the half-wave potential of Fe (CN)63−/4− pair and MV2+ considerably, which thus lowers the energy barrier for the occurrence of electrochemical reaction. This is a general effect regardless of the sign of charge(s) carried by the redox active species. However, the promotion of current may occur to positively charged and neutral ones owing to the enhanced electroattractive interaction between the film and the probes. The current might be considerably decreased in cases of negatively charged species due to the repulsive impedance caused by the accumulated electrons in the reducing process. Our results demonstrate a representative case of the catalytic properties of soft coordination polymers. This can be applied to the detection of physiologically important substances such as dopamine and hydrogen peroxide, and may open a new vista towards function-directed rational design of coordination supramolecular polymers and may shed light on advanced catalytic materials.Acknowledgements
This work was part of projects 20903005, 21173011, 20805002, and 21073006, supported by National Natural Science Foundation of China (NSFC).References
- G. R. Whittell, M. D. Hager, U. S. Schubert and I. Manners, Nat. Mater., 2011, 10, 176 CrossRef CAS.
- J. S. Kim, H. An, W. J. Rieter, D. Esserman, K. M. L. Taylor-Pashow, R. B. Sartor, W. Lin, W. Lin and T. K. Tarrant, Clinical and Experimental Rheumatology, 2009, 27, 580 CAS.
- A. M. Spokoyny, D. Kim, A. Sumrein and C. A. Mirkin, Chem. Soc. Rev., 2009, 38, 1218 RSC.
- X. J. Zhang, M. A. Ballem, M. Ahren, A. Suska, P. Bergman and K. Uvdal, J. Am. Chem. Soc., 2010, 132, 10391 CrossRef CAS.
- X. J. Zhang, M. A. Ballem, Z. J. Hu, P. Bergman and K. Uvdal, Angew. Chem., Int. Ed., 2011, 50, 5729 CrossRef CAS.
- J. B. Beck and S. J. Rowan, J. Am. Chem. Soc., 2003, 125, 13922 CrossRef CAS.
- A. M. S. Kumar, S. Sivakova, J. D. Fox, J. E. Green, R. E. Marchant and S. J. Rowan, J. Am. Chem. Soc., 2008, 130, 1466 CrossRef CAS.
- S. Burattini, B. W. Greenland, D. H. Merino, W. G. Weng, J. Seppala, H. M. Colquhoun, W. Hayes, M. E. Mackay, I. W. Hamley and S. J. Rowan, J. Am. Chem. Soc., 2010, 132, 12051 CrossRef CAS.
- J. R. Kumpfer and S. J. Rowan, J. Am. Chem. Soc., 2011, 133, 12866 CrossRef CAS.
- T. Vermonden, M. J. van Steenbergen, N. A. M. Besseling, A. T. M. Marcelis, W. E. Hennink, E. J. R. Sudholter and M. A. C. Stuart, J. Am. Chem. Soc., 2004, 126, 15802 CrossRef CAS.
- M. Burnworth, L. M. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Nature, 2011, 472, 334 CrossRef CAS.
- D. G. Kurth and M. Higuchi, Soft Matter, 2006, 2, 915 RSC.
- Y. Yan and J. B. Huang, Coord. Chem. Rev., 2010, 254, 1072 CrossRef CAS.
- S. J. Rowan and J. B. Beck, Faraday Discuss., 2005, 128, 43 RSC.
- D. Knapton, M. Burnworth, S. J. Rowan and C. Weder, Angew. Chem., Int. Ed., 2006, 45, 5825 CrossRef CAS.
- J. R. Kumpfer, J. Z. Jin and S. J. Rowan, J. Mater. Chem., 2010, 20, 145 RSC.
- F. S. Han, M. Higuchi, T. Ikeda, Y. Negishi, T. Tsukuda and D. G. Kurth, J. Mater. Chem., 2008, 18, 4555 RSC.
- L. Yang, Y. Ding, Y. Yang, Y. Yan, J. B. Huang, A. de Keizer and M. A. Cohen Stuart, Soft Matter, 2011, 7, 2720 RSC.
- Y. Bodenthin, G. Schwarz, Z. Tomkowicz, M. Lommel, T. Geue, W. Haase, H. Mohwald, U. Pietsch and D. G. Kurth, Coord. Chem. Rev., 2009, 253, 2414 CrossRef CAS.
- Y. Yan, Y. R. Lan, A. de Keizer, M. Drechsler, H. Van As, M. A. C. Stuart and N. A. M. Besseling, Soft Matter, 2010, 6, 3244 RSC.
- Y. Ding, Y. Yang, L. Yang, Y. Yan, J. B. Huang and M. A. Cohen Stuart, ACS Nano, 2012, 6, 1004 CrossRef CAS.
- L. Zhao, Y. Yan and J. B. Huang, Langmuir, 2012, 28, 5548 CrossRef CAS.
- L. Q. Ma, C. Abney and W. B. Lin, Chem. Soc. Rev., 2009, 38, 1248 RSC.
- S. Q. Liu, D. Volkmer and D. G. Kurth, J. Cluster Sci., 2003, 14, 405 CrossRef CAS.
- R. R. Pal, M. Higuchi, Y. Negishi, T. Tsukuda and D. G. Kurth, Polym. J., 2010, 42, 336 CrossRef CAS.
- D. G. Kurth and M. Schutte, Colloids Surf., A, 2002, 198, 633 CrossRef.
- J. B. Schlenoff, D. Laurent, H. Ly and J. Stepp, Adv. Mater., 1998, 10, 347 CrossRef CAS.
- J. Y. Wang, A. de Keizer, R. Fokkink, Y. Yan, M. A. C. Stuart and J. van der Gucht, J. Phys. Chem. B, 2010, 114, 8313 CrossRef CAS.
- J. Y. Wang, A. de Keizer, H. P. Van Leeuwen, Y. Yan, F. Vergeldt, H. Van As, P. H. H. Bomans, N. A. J. M. Sommerdijk, M. A. Cohen Stuart and J. van der Gucht, Langmuir, 2011, 27, 14776 CrossRef CAS.
- Y. Yan, N. A. M. Besseling, A. de Keizer, A. T. M. Marcelis, M. Drechsler and M. A. Cohen Stuart, Angew. Chem., Int. Ed., 2007, 46, 1807 CrossRef CAS.
- Y. Yan, A. A. Martens, N.A.M. Besseling, F. A. de Wolf, A. de Keizer, M. Drechsler and M. A. Cohen Stuart, Angew. Chem., Int. Ed., 2008, 47, 4192 CrossRef CAS.
- Y. R. Lan, L. M. Xu, Y. Yan, Y. Yan, J. B. Huang, A. de Keizer, N. A. M. Besseling and M. A. Cohen Stuart, Soft Matter, 2011, 7, 3565 RSC.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods Fundamentals and Applications, John Wiley & Sons, 2nd Edition. 2001 Search PubMed.
- D.G. Kurth and R. Osterhout, Langmuir, 1999, 15, 4842 CrossRef CAS.
- J. J. Harris and M. L. Bruening, Langmuir, 2000, 16, 2006 CrossRef CAS.
- V. P. Menon and C. R. Martin, Anal. Chem., 1995, 67, 1920 CrossRef CAS.
- O. Chailapakul and R. M. Crooks, Langmuir, 1995, 11, 1329 CrossRef CAS.
- E. E. Engelman and D. H. Evans, Langmuir, 1992, 8, 1637 CrossRef CAS.
- Y. T an, W. Deng, Y. Li, Z. Huang, Y. Meng, Q. Xie, M. Ma and S. Yao, J. Phys. Chem. B, 2010, 114, 5016 CrossRef CAS.
- L. Wang and E. Wang, Electrochem. Commun., 2004, 6, 225 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ra22237j |
| ‡ These authors equally contributed to this work |
| This journal is © The Royal Society of Chemistry 2012 |