Lyotropic liquid crystalline phases formed in ternary mixtures of N-alkyl-N-methylpyrrolidinium bromide/1-decanol/water†
Lijuan
Shi
a,
Mingwei
Zhao
b and
Liqiang
Zheng
*a
aKey Laboratory of Colloid and Interface Chemistry, Shandong University, Ministry of Education, Jinan, Shandong, 250100, China. E-mail: lqzheng@sdu.edu.cn; Fax: +86-531-88564750; Tel: +86-531-88366062
bCollege of Petroleum Engineering, China University of Petroleum (East China), Qingdao, Shandong, 266555, China
First published on 5th October 2012
Abstract
The phase behavior of ternary systems involving long-chain pyrrolidinium ionic liquids, N-alkyl-N-methylpyrrolidinium bromide (CnMPB, n = 12, 14, 16), water, and 1-decanol was investigated at 25 °C. Polarized optical microscopy (POM), small-angle X-ray scattering (SAXS), and rheological measurements were employed to investigate the lyotropic liquid crystalline (LC) phases. Lamellar phase (Lα) and hexagonal phase (H1) were found to exist in all the three systems, but the isotropic cubic phase only appears in the C12MPB and C14MPB systems. Greater surfactant content leads to a denser aggregation of the cylindrical units in the H1 phase, and the structural parameters of Lα phase depends on water content. The alkyl chain length of CnMPB also has interesting effect on the structural parameters and rheological properties of the LC phase. Compared with a similar ternary system of 1-hexadecyl-3-methylimidazolium chloride ([C16mim]Cl) and traditional cationic surfactants cetyltrimethylammonium bromide (C16TAB), the effect of the cationic group on the phase behavior was also investigated.
1. Introduction
Ionic liquids (ILs) have significantly developed recently because of their extraordinary properties, such as nonvolatility, nonflammability, high stability, high ionic conductivity, and easy recyclability.1,2 Numerous ILs based on imidazolium, pyrrolidinium, and pyridinium cations with a variety of anions have been successively synthesized and widely utilized in organic synthesis, catalysis, and the preparation of nanostructured materials.3–5 An interesting characteristic of ILs is that their cations, like the imidazolium, pyrrolidinium, pyridinium, and quaternary ammonium, are inherently amphiphilic. Therefore, ILs with long alkyl chain can also be regarded as novel kinds of amphiphiles to form aggregates with specific structure, shape and properties.Generally, amphiphile molecules can form thermodynamically stable self-organized structures, such as micelles, microemulsions, vesicles, and liquid crystals (LCs).6–9 Among them, LCs have received much attention due to their wide applications in chemical reactions, material science, and pharmaceutical vehicles.10–13 Recently, LC phases formed by IL-typed surfactants have been studied extensively. Firestone and co-workers have found that LC gels can be formed by the 1-decyl-3-methylimidazolium bromine ([C10mim]Br) and 1-decyl-3-methylimidazolium nitrate ([C10mim]NO3) in aqueous solution.14–16 Smarsly and co-workers have clarified the LC phases in the [C16mim]Cl–H2O binary system.17,18 Goodchild et al. have performed 2H NMR spectroscopy measurements to confirm the formation of a lyotropic mesophase in the [C8mim]Cl–H2O system.19 Lamellar and hexagonal phases in the [C12mim]Br–H2O system have been presented by Inoue et al. through polarized optical microscopy (POM) and differential scanning calorimetry (DSC).20 Ternary LC systems concerning surface active [Cnmim]X salts with water and oil have also been observed. Chen et al. have reported the LC phase formed by [C16mim]Cl, with 1-decanol and water.21 The hydrogen-bonded network comprised of the imidazolium ring, anion, 1-decanol, and water plays important roles in the formation of LC phase. Then they investigated the effect of carbon chain length and content of alcohols on the phase behavior, finding that the strong interaction between counterions and alcohols favors the appearance of ordered assemblies.22 The LC phases formed in the [Cnmim]Br-p-xylene-water system have been studied by our group.23,24 The unique properties of [Cnmim]Br, such as “π–π stacking” and “π-cation” interactions, play an important role in determining the internal structural parameters. All the research above suggests that a suitable molecule design could subtly tune the phase behavior of surfactants. However, most of the research has been confined to imidazolium ILs. Very recently, research has expanded to ILs with other cations, like pyrrolidinium. We have reported LC phases formed by pyrrolidinium ILs, N-hexadecyl-N-methylpyrrolidinium bromide (C16MPB) in water and a room temperature IL ethylammonium nitrate (EAN).25
As an extension of these preceding studies, we investigated the phase behavior of ternary systems involving N-alkyl-N-methylpyrrolidinium bromide (CnMPB, n = 12, 14 and 16) with 1-decanol and H2O for the first time in the present work. The phase behavior was studied at 25 °C with a particular focus on the liquid crystal regions using SAXS, POM, and rheology measurements. The effect of the alkyl chain length of CnMPB on the phase behavior was systematically investigated. Compared with a similar ternary system of the imidazolium IL [C16mim]Cl and the conventional cationic surfactant C16TAB, the effect of the cationic group on the phase behavior was also investigated. We expect that the present study is helpful to better understand the effect of structure on the phase behavior of ILs.
2. Experimental section
2.1 Materials
N-Methylpyrrolidine (97%), 1-bromododecane (97%), 1-bromotetradecane (97%), 1-bromohexadecane(97%), and 1-decanol were purchased from Sigma. Toluene (99%) and diethyl ether (99%) were purchased from Beijing Chemical Reagent Company. Deionized water was used through the experiment.2.2 Phase diagram
Samples for mapping phase diagrams were prepared by weighing all components at designed compositions (in weight percent, wt%). CnMPB and 1-decanol were mixed with their weight ratio varying from 0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 to 10:0, and the water content was selected along the water dilute line. The mixtures in all systems were homogenized and equilibrated by repeated vortex mixing and centrifugation. Then the samples were kept at 25 °C for at least 1 month before further examination. The types of liquid crystals were identified by polarizing light microscopy (POM) and small-angle X-ray scattering (SAXS). The experimental samples with different morphologies are shaded in different symbols in the phase diagrams as shown in Fig. 1.
:10 to 10:0, and the water content was selected along the water dilute line. The mixtures in all systems were homogenized and equilibrated by repeated vortex mixing and centrifugation. Then the samples were kept at 25 °C for at least 1 month before further examination. The types of liquid crystals were identified by polarizing light microscopy (POM) and small-angle X-ray scattering (SAXS). The experimental samples with different morphologies are shaded in different symbols in the phase diagrams as shown in Fig. 1.
 | ||
Fig. 1 Ternary phase diagrams for C12MPB–C10H21OH–H2O (A), C14MPB–C10H21OH–H2O (B), and C16MPB–C10H21OH–H2O (C) systems at 25 °C. Isotropic solutions (●, ); lamellar liquid crystalline phase (■); hexagonal liquid crystalline phase ( ); lamellar liquid crystalline phase (■); hexagonal liquid crystalline phase ( ); cubic liquid crystalline phase (▲). ); cubic liquid crystalline phase (▲). | ||
2.3 Characterization
A polarized optical microscope (Olympus BX51p) equipped with cooled CCD (Evolution MP5.1RTV, Q-imaging, Canada) was used to observe textures of the LC phase. The temperature was controlled at 25 °C with a Linkam THSME600 liquid crystal freezing and heating stage system with a TP94 temperature controller (Linkam Scientific Instrument Ltd., UK).SAXS measurements were performed using a Kratky Compact Camera (HMBG, Austria) with Ni and W filtered Cu-Kα radiation (wavelength λ = 0.15418 nm) generated by a PW3830 X-ray generator (5 kV × 40 mA). Scattering intensities were plotted versus reciprocal spacing (q = 4πsinθ/λ), where θ was the scattering angle. The exposure time was 600 s for all samples.
Rheology measurements were performed with a HAAKE RS 75 rheometer. A cone-plate sensor was used with 20 mm diameter and 1° cone angle. The cone-plate distance was adjusted to 52 μm for all measurements. The temperature was kept at 25 ± 0.1 °C.
3. Results and discussion
3.1 Phase behaviors of ternary systems
Ternary mixtures of CnMPB with different alkyl chain length, H2O and 1-decanol were investigated at 25 °C. Their phase diagrams are shown in Fig. 1. For all the systems, single-phase region consists of the isotropic solution phase, anisotropic lamellar phase (Lα) and hexagonal phase (H1). It is interesting to find that isotropic cubic phase exists in the C12MPB and C14MPB systems but disappears when the alkyl chain length is increased to 16. The LC phases were determined by a combination of POM and SAXS measurements.
Fig. 2 shows the POM images and corresponding SAXS curves of three different liquid crystal samples in the C12MPB–C10H21OH–H2O ternary system. The POM image in Fig. 2 (A) exhibits the coexistence of crossed textures and oiled veins, which is the typical texture for the Lα phase. Two Bragg peaks in the scattering curve with the relative positions of 1:2 further indicates a lamellar structure. A characteristic “fan” pattern for the hexagonal lattice is illustrated in Fig. 2 (B). The corresponding SAXS spectrum exhibits the Bragg peaks with the ratio of 1:√3, indicating that the allowed reflections are from the hexagonal symmetry group. As shown in Fig. 2 (C), no birefringence is observed through a crossed polarizer, indicating that this sample is isotropic. The Bragg peaks in the SAXS spectrum can be indexed as the (211) and (220) reflections of the Ia3d structure, with the ratio q1:q2 = √3:√4. Similar result can also be observed in the C14MPB–C10H21OH–H2O ternary system. However, we did not observe the existence of the isotropic cubic phase in the C16MPB–C10H21OH–H2O system.
 | ||
| Fig. 2 POM images (top) and SAXS curves (bottom) for the typical liquid crystal phases: (A) Lα, (B)H1, and (C) C in the C12MPB–C10H21OH–H2O system. | ||
The incorporation of 1-decanol into the CnMPB–H2O systems brings interesting changes of the phase behavior. As reported previously, only H1 phase were observed in the C16MPB–H2O binary system at the room temperature.25 With the addition of 1-decanol, a large Lα phase region appears. In general, the oil can be incorporated into a surfactant aggregate through two ways: “penetration,” in which the oil acts as a co-surfactant whose headgroup exists between adjacent surfactant molecules at the aggregate interface; and “swelling,” in which the oil acts as a co-solvent and is located in the core of the aggregate.27,28 The incorporated C10H21OH in the CnMPB–H2O aggregates could act as both the co-solvent and co-surfactant.21 The presence of C10H21OH reduces the aggregate curvature in the LC phases and then leads to a phase transition from hexagonal to lamellar.29 The possible packing model in the H1 and Lα phases of the CnMPB–C10H21OH–H2O ternary systems are therefore drawn as shown in Scheme 1.
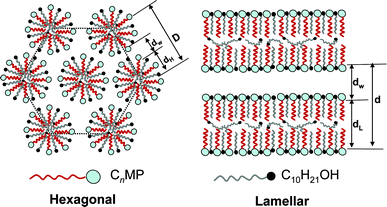 | ||
| Scheme 1 Sectional schematic graph of the possible structures formed in the H1 and Lα phases of the CnMPB–C10H21OH–H2O ternary systems. | ||
It is clear that the change of alkyl chain length can cause some differences in the phase behavior of the CnMPB–C10H21OH–H2O system. As the carbon chain increases in length, the liquid crystalline region moves closer to the water corner, indicating that the formation of the liquid crystal become somewhat easier. The explanation might be ascribed to the decreased hydrophilicity of the CnMPB, resulting in weaker hydration ability.30,31 In addition, the result can also be explained from the viewpoint of the critical packing parameter (CPP). The CPP is determined as P = V/(al), where P is the abbreviation of CPP, V is the volume of the solvophobic chains of surfactant molecules, a is the effective headgroup area of the surfactant molecules, and l is the effective chain length of surfactant molecules. In general, the values of CPP are as follows: for spherical micelles, P < ⅓; for cylindrical aggregates, ⅓ < P < ½; for bilayers, vesicles, and bicontinuous cubic LC phase, ½ < P <1; and for inverted structures, P > 1. For the CnMPB molecules, the hydrocarbon chain length l and volume of the solvophobic chains V can be obtained from the following Tanford equations:32
| l = 1.5 + 1.265N | (1) |
| V = 27.4 + 26.9N | (2) |
Comparing the phase behavior of the C16MPB–C10H21OH–H2O system with that of the [C16mim]Cl–C10H21OH–H2O and C16TAB–C10H21OH–H2O systems, the effect of headgroups on the phase behavior was also investigated. The typical phase diagrams for C16TAB–C10H21OH–H2O29 and [C16mim]Cl–C10H21OH–H2O21 are shown in Fig. S1, ESI†. It is clear that all these systems show the same sequence of phases (L1, H1, Lα, L2) upon addition of C10H21OH. However, some dissimilarities are also present in the three ternary systems. Firstly, in the C16TAB system, the reversed lyotropic phases such as the reversed hexagonal phase (F) and another anisotropic phase with a non-hexagonal inverted rod-aggregates in a non-polar hydrocarbon environment (K), can be discovered. Secondly, the H1 phase in the C16MPB system can accommodate higher concentrations (6 wt%) of C10H21OH, while only about 2 wt% C10H21OH can be solubilized in the C16TAB system. In addition, the Lα phase in the C16MPB system covers a much more narrow water concentration range (20–45 wt%) than the Lα phase in the C16TAB system (25–96 wt%) and [C16mim]Cl system (5–85 wt%). As reported previously, the capability to accommodate water in the systems containing cationic surfactant and alcohol is related to the nature of the counterions and the chain length of the alcohol.17,29 The present result further indicates that the headgroup of a surfactant is another factor to influence the capability to accommodate water in the cationic surfactant-alcohol-water systems.
Since C16MPB and C16TAB have the same alkyl chain length and counterion, the change in the phase behaviors of the C16MPB–C10H21OH–H2O and C16TAB–C10H21OH–H2O systems is mainly caused by the difference in head groups. The difference in the head groups causes different spontaneous curvature of the surfactant monolayer (H0) and degree of counterion binding (β). The spontaneous curvature of the surfactant monolayer (H0), which indicates the curvature free energy, is determined by a variety of interactions between the membranes.33–36 In ionic surfactant systems, besides the long-range Helfrich repulsion caused by thermal undulations of the flexible bilayers, the electrostatic interaction between head groups is another important factor to stabilize the surfactant membranes. As the electrostatic interactions between the head groups increase, H0 is driven towards less negative values.34 As reported previously, the degree of counterion binding for CnMPB is lower than that for CnTAB with the same alkyl chain length.37 This result means that the self-repulsion of the headgroups for C16MPB is stronger than that for C16TAB, resulting in much weaker electrostatic interactions between the headgroups for C16MPB compared with C16TAB. Thus, the H0 value for surfactant membranes in the C16MPB system would be more negative than C16TAB. Consequently, it is more difficult for the hexagonal phase in the C16MPB system to be transformed to the lamellar phase and reversed lyotropic phases compared with the C16TAB systems. That is, more C10H21OH is needed to transform the hexagonal phase in the C16TAB system to lamellar phase compared with the C16TAB systems. In addition, the more negative H0 value in the C16MPB system compared with C16TAB can also be used to explain the lack of reversed LLC phases in the C16MPB system.
3.2 Structural parameters of the liquid crystalline phase
The SAXS characterization of the LC phase is based on the long range order in the LC state, which leads to the Bragg reflections. The positions of these Bragg reflections are characteristic of the different types of LC phases, and the relevant structural parameters can be deduced from the SAXS pattern. Fig. 3 shows the SAXS patterns for the representative samples of H1 phase (A) and Lα phase (B) constructed in the C12MPB–C10H21OH–H2O system at 25 °C. All of the parameters calculated from the SAXS results were also obtained according to the equations and theory shown in the supporting information, and the values are listed in Table S1, ESI.† | ||
| Fig. 3 SAXS spectra of samples in hexagonal (A) and lamellar (B) phases in the C12MPB–C10H21OH–H2O system. The compositions of the samples (C12MPB–C10H21OH–H2O, mol%) are shown in the figure. | ||
Fig. 4 shows the variation of the thickness of the water channel (dw) and the radius of the cylinder unit (dH) as the function of the mole fraction of CnMPB (ϕs) in the hexagonal phase. In the C12MPB system, it can be seen that the values of dw decrease, and dH increase as the C12MPB content increases. In other words, the radius of the cylinder-like aggregates becomes larger with the increasing amount of C12MPB, while the solvent layer becomes thinner. This result suggests that at higher C12MPB concentration, the cylinder-like aggregates pack more densely in the hexagonal phase, but the arrangement of surfactant molecules per cylinder-like aggregate becomes looser. Similar result can also be observed in the C14MPB system. However, it is interesting that a different tendency was observed in the C16MPB system. As the C16MPB content increases, both the values of dw and dH decrease, indicating that the cylinder-like aggregates pack more densely in the hexagonal phase and the size of the cylinder-like aggregates decreases. This result may be due to the fact that the hydrophobic chain of C16MPB shrinks more in the hexagonal region containing more surfactants.
 | ||
| Fig. 4 Dependence of the thickness of the water channel (dw) and the radius of the cylinder unit (dH) on the mole fraction of CnMPB (ϕs) for the hexagonal phase in the CnMPB–C10H21OH–H2O (n = 12, 14, 16) systems with fixed mole ratio of decanol to H2O = 1:150. | ||
Fig. 5 shows the variation of the thickness of hydrophobic domain (dL), and the area per polar group at the hydrophilic/hydrophobic interface (as) as the function of the mole fraction of water (ϕw) in the lamellar phase. It can be seen that when the ratio of the mole ratio of 1-decanol:C12MPB is fixed, dL decreases with increasing water content. This result means that as the water content in the lamellar phase increases, the surfactant bilayer is compressed by the expanding water channel. In contrast, the as value of C12MPB was found to increase with increasing water content. This phenomenon coincides with the general phenomenon for ionic surfactants that the headgroup area depends on the water content.38,39 The results for the C14MPB and C16MPB systems are similar to the C12MPB system. However, the change of alkyl chain length can also bring some differences about the structural parameters. As shown in Fig. 5, the value of dL increases as the alkyl chain length is increased, while the value of as is independent of the number of carbons in the hydrophobic group.
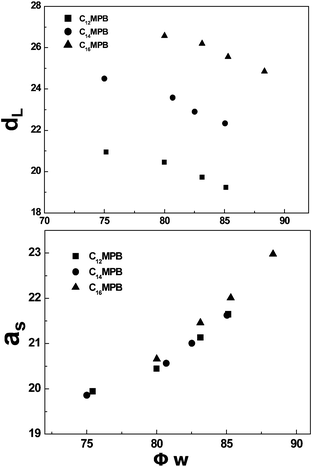 | ||
| Fig. 5 Dependence of the thickness of hydrophobic domain (dL) and the area per polar group at the hydrophilic/hydrophobic interface (as) on the mole fraction of water (ϕw) for the lamellar phase in the CnMPB–C10H21OH–H2O (n = 12, 14, 16) systems with fixed mole ratio of decanol to surfactant = 1:2.5. | ||
3.3 Rheological properties of the liquid crystalline phase
More information on the network structure of the liquid crystalline phase was obtained from rheological measurements. Fig. 6 shows the dynamic viscoelastic behavior for the representative samples of H1 phase (A) and Lα phase (B) constructed in the C12MPB–C10H21OH–H2O system at 25 °C. The sample compositions in the H1 phase (H1-1, H1-2, and H1-3) and in the Lα phase (Lα-1, Lα-2, and Lα-3) are the same as those shown in Fig. 3. As shown in Fig. 6 (A), the values of the storage modulus (G′) increase versus the frequency, while the loss modulus (G′′) increases with increasing frequency at the beginning and then remains almost constant. At low frequencies, the loss modulus G′′ is larger than the storage modulus G′, showing viscous behavior. Then G′ becomes larger than G′′ at higher frequencies, exhibiting elastic behavior. The rheological results mean that the H1 phase shows viscoelastic behavior, which is consistent with the typical rheological behavior of H1 phases.40 The reciprocal of the frequency corresponding to the intersection point of G′ and G′′ is defined as the relaxation time, τ.41 A larger value of τ means a longer exchange time for the surfactant molecules between the cylinder unit and aqueous phase. The obtained τ values of the samples in Fig. 6 (A) are 15.73, 19.84, and 28.66s respectively, meaning that exchange time for the surfactant molecules between the cylinder unit and aqueous phase increases with increasing C12MPB content. That is, the diffusion of the surfactant molecules between the cylinder unit and aqueous phase becomes slower as the C12MPB content increases. For the lamellar phase shown in Fig. 6 (B), the storage modulus G′ is higher than the loss modulus G′′ over the whole frequency range, indicating that the phases correspond to a “weak gel”.42 Meanwhile, the values of dynamic moduli are practically independent of the frequency, and these properties are in agreement with those of lamellar liquid crystalline phase formed from traditional surfactants.43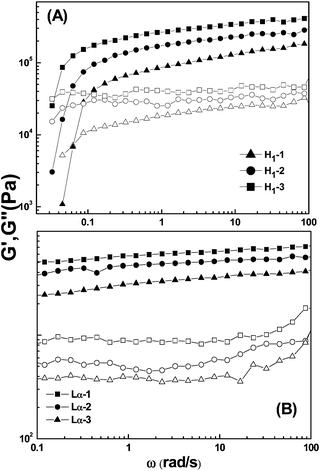 | ||
| Fig. 6 Storage moduli (G′, solid) and loss moduli (G′′, hollow) as a function of angular frequency (ω) for hexagonal (A) and lamellar (B) phases in the C12MPB–C10H21OH–H2O system. The compositions of the samples are as those shown in Fig. 3. | ||
The stable flow behavior curves of the LC phases in the C12MPB–C10H21OH–H2O system are shown in Fig. 7. All of the samples behave as shear-thinning fluids, and the viscosities of the hexagonal phases are much higher than the viscosities of the lamellar phases. The zero-shear-rate viscosity (η0) could be obtained by extrapolating the viscosity against the shear rate curves back to the viscosity intercept at zero shear rate. It can be seen in the hexagonal phase, the η0 value increases with increasing surfactant content, and this is due to the much denser cylinder-like aggregates in the hexagonal phase at higher surfactant content. On the contrary, a decrease in η0 is observed upon addition of water in the lamellar phase. This result also coincides with the changes of the structural parameters in the lamellar phase.
 | ||
| Fig. 7 Viscosity (η) versus shear rate for hexagonal (A) and lamellar (B) phases in the C12MPB–C10H21OH–H2O system. The compositions of the samples are as those shown in Fig. 3. | ||
The effect of the carbon chain on the rheological properties of the liquid crystalline phases was also investigated. Fig. 8 shows the dynamic viscoelastic behavior for the samples with the same component content in H1 phase (A) and Lα phase (B) for CnMPB (n = 12, 14, 16). The sample compositions (CnMPB–C10H21OH–H2O, mol%) were chosen to be about 6.46–0.65–92.89% in the H1 phase, and about 11.16–3.73–85.11% in the Lα phase. It is found that in the H1 phase, the storage modulus G′ increases with increasing carbon chain length of CnMPB. The tendency is similar to traditional surfactant/oil/water ternary systems.44 As shown in Fig. 9 (A), the zero-shear-rate viscosity η0 for the samples in H1 phase is also increased as the carbon chain becomes longer. However, the loss modulus G′′ in the C14MPB system is larger than in the C12MPB system at low frequencies but becomes somewhat smaller than C12MPB at higher frequencies. The difference may be due to the fact that the H1 phase exhibits typical elastic behavior at higher frequencies. As a result, the changes of viscous modulus G′′ may be not as obvious and typical as the elastic modulus G′ at higher frequencies. In addition, the structural parameters for the samples in the H1 phase have similar changing trend as the C12MPB and C14MPB content increases, but the opposite change was observed in the C16MPB system. The phase behaviors of C12MPB and C14MPB in other systems are also found to be different with C16MPB.45 The phenomenon above indicates that the phase behaviors of C12MPB and C14MPB under the same conditions are very similar. This is also perhaps the reason that the dynamic viscoelastic behavior for the samples with the same component content in H1 phase in the C12MPB and C14MPB systems does not change typically.
 | ||
| Fig. 8 Storage moduli (G′, solid) and loss moduli (G′′, hollow) as a function of angular frequency (ω) for hexagonal (A) and lamellar (B) phases for the samples with the same component content for CnMPB (n = 12, 14, 16). The compositions of the samples (CnMPB–C10H21OH–H2O, mol%) are 6.46–0.65–92.89% (A) and 11.16–3.73–85.11% (B). | ||
 | ||
| Fig. 9 Viscosity (η) versus shear rate for hexagonal (A) and lamellar (B) liquid crystalline phases for the samples with the same component content for CnMPB (n = 12, 14, 16). The compositions of the samples (CnMPB–C10H21OH–H2O, mol%) are 6.46–0.65–92.89% (A) and 11.16–3.73–85.11% (B). | ||
However, the carbon chain has different effect on the rheological properties of the Lα phases. As shown in Fig. 8 (B), the rheological steady and dynamic moduli decrease slightly with the increased carbon chain length of CnMPB. And the zero-shear-rate viscosity η0 is also decreased with increasing carbon chain length. This result may be due to the increased dW values as the carbon chain length increased.
Conclusion
In the ternary systems formed by CnMPB (n = 12, 14, 16) with water and 1-decanol, the lamellar phase (Lα) and hexagonal phase (H1) were found to be formed. The cubic phase also appears only in the C12MPB and C14MPB systems. Greater surfactant content leads to denser aggregation of the cylindrical units in the H1 phase. In the Lα phase, the increase in the lattice parameter and thickness of the water layer is attributed to the increase of water content. The structural parameters and rheological properties of the LC phase are also affected by the change of surfactant alkyl chain length. The size of the cylinder-like aggregates in the H1 phase becomes larger as the C12MPB and C14MPB content increases, but the opposite change was observed in the C16MPB system. The thickness of the hydrophobic domain and water layer in the Lα phase increases with increasing alkyl chain length, while the area per surfactant molecule at the hydrophobic/hydrophilic interface keeps almost unchanged. In the H1 phase, the storage modulus increases with increased carbon chain length of CnMPB, while the change in loss modulus is not regular. Slight decreases for both the storage and loss moduli in the Lα phase are observed with increased carbon chain length of CnMPB. In addition, the change of the headgroups could also cause some differences in the LC behavior of cationic surfactants.Acknowledgements
The authors are grateful to the National Basic Research Program (2013CB834505), the National Natural Science Foundation of China (No. 91127017), and the Shandong Provincial Natural Science Foundation, China (ZR2012BZ001) for financial support.References
- R. D. Rogers and K. R. Seddon, Science, 2003, 302, 792–793 CrossRef.
- T. Welton, Chem. Rev., 1999, 99, 2071–2084 CrossRef CAS.
- C. E. Song, Chem. Commun., 2004, 1033–1043 RSC.
- D. B. Zhao, M. Wu, Y. Kou and E. Z. Min, Catal. Today, 2002, 74, 157–189 CrossRef CAS.
- Y. Wang and H. Yang, J. Am. Chem. Soc., 2005, 127, 5316–5317 CrossRef CAS.
- M. Abe, H. Uchiyama, T. Yamaguchi, T. Suzuki and K. Ogino, Langmuir, 1992, 8, 2147–2151 CrossRef CAS.
- J. H. Schulman, W. Stoeckenius and L. M. Prince, J. Phys. Chem., 1959, 63, 1677–1680 CrossRef CAS.
- E. W. Kaler, A. K. Murthy, B. E. Rodriguez and J. A. N. Zasadzinski, Science, 1989, 245, 1371–1374 CAS.
- J. W. Park, C. S. Bak and M. M. Labes, J. Am. Chem. Soc., 1975, 97, 4398–4400 CrossRef CAS.
- A. Taubert, Angew. Chem., Int. Ed., 2004, 43, 5380–5382 CrossRef CAS.
- D. L. Gin, W. Q. Gu, B. A. Pindzola and W. J. Zhou, Acc. Chem. Res., 2001, 34, 973–980 CrossRef CAS.
- W. Torres and M. A. Fox, Chem. Mater., 1992, 4, 583–588 CrossRef CAS.
- D. I. Nesseem, J. Pharm. Biomed. Anal., 2001, 26, 387–399 CrossRef CAS.
- M. A. Firestone, J. A. Dzielawa, P. Zapol, L. A. Curtiss, S. Seifert and M. L. Dietz, Langmuir, 2002, 18, 7258–7260 CrossRef CAS.
- M. A. Firestone, P. G. Rickert, S. Seifert and M. L. Dietz, Inorg. Chim. Acta, 2004, 357, 3991–3998 CrossRef CAS.
- M. A. Firestone, M. L. Dietz, S. Seifert, S. Trasobares, D. J. Miller and N. J. Zaluzec, Small, 2005, 1, 754, 25–27 Search PubMed.
- H. Kaper and B. Z. Smarsly, Phys. Chem., 2006, 220, 1455–1471 CAS.
- T. W. Wang, H. Kaper, M. Antonietti and B. Smarsly, Langmuir, 2007, 23, 1489 CrossRef CAS.
- I. Goodchild, L. Collier, S. L. Millar, I. Prokes, J. C. D. Lord, C. P. Butts, J. Bowers, J. R. P. Webster and R. K. Heenan, J. Colloid Interface Sci., 2007, 307, 455–468 CrossRef CAS.
- T. Inoue, B. Dong and L. Q. Zheng, J. Colloid Interface Sci., 2007, 307, 578–581 CrossRef CAS.
- G. D. Zhang, X. Chen, Y. Z. Xie, Y. R. Zhao and H. Y. Qiu, J. Colloid Interface Sci., 2007, 315, 601–606 CrossRef CAS.
- G. Zhang, X. Chen, Y. Zhao and H. Qiu, J. Phys. Chem. B, 2007, 111, 11708–11713 CrossRef CAS.
- J. Zhang, B. Dong, L. Q. Zheng, N. Li and X. W. Li, J. Colloid Interface Sci., 2008, 321, 159–165 CrossRef CAS.
- X. W. Li, J. Zhang, B. Dong, L. Q. Zheng and C. H. Tung, Colloids Surf., A, 2009, 335, 80–87 CrossRef CAS.
- M. W. Zhao, Y. A. Gao and L. Q. Zheng, J. Phys. Chem. B, 2010, 114, 11382–11389 CrossRef CAS.
- K. Goossens, K. Lava, P. Nockemann, K. Van Hecke, L. Van Meervelt, K. Driesen, C. Görller-Walrand, K. Binnemans and T. Cardinaels, Chem.–Eur. J., 2009, 15, 656–674 CrossRef CAS.
- J. S. Bernardes, J. Norrman, L. Piculell and W. Loh, J. Phys. Chem. B, 2006, 110, 23433–23442 CrossRef CAS.
- H. Kunieda, K. Ozawa and K. L. Huang, J. Phys. Chem. B, 1998, 102, 831–838 CrossRef CAS.
- K. Fontell, A. Khan, B. Lindstrom, D. Maciejewska and S. Puang-Ngern, Colloid Polym. Sci., 1991, 269, 727–742 CAS.
- L. K. Shrestha, M. Kaneko, T. Sato, D. P. Acharya, T. Iwanaga and H. Kunieda, Langmuir, 2006, 22, 1449–1454 CrossRef CAS.
- A. Ishikawa, K. Tamori, K. Esumi and K. Meguro, J. Colloid Interface Sci., 1992, 151, 370–382 CrossRef CAS.
- C. Tanford, J. Phys. Chem., 1972, 76, 3020–3024 CrossRef CAS.
- J. Gustafsson, G. Orädd, G. Lindblom, U. Olsson and M. Almgren, Langmuir, 1997, 13, 852–860 CrossRef CAS.
- E. Pena dos Santos, M. S. Tokumoto, G. Surendran, H. Remita, C. Bourgaux, P. Dieudonne, E. Prouzet and L. Ramos, Langmuir, 2005, 21, 4362–4369 CrossRef.
- A. Zou, H. Hoffmann, N. Freiberger and O. Glatter, Langmuir, 2007, 23, 2977–2984 CrossRef CAS.
- P. D. Galgano and O. A. El Seoud, J. Colloid Interface Sci., 2010, 345, 1–11 CrossRef CAS.
- M. Tariq, A. Podgoršek, J. L. Ferguson, A. Lopes, M. F. Costa Gomes, A. A. H. Pádua, L. P. N. Rebelo and J. N. Canongia Lopes, J. Colloid Interface Sci., 2011, 360, 606–616 CrossRef CAS.
- H. Edlund, A. Sadaghiani and A. Khan, Langmuir, 1997, 13, 4953–4963 CrossRef CAS.
- K. Fontell, L. Mandell, H. Lehtinen and P. Ekwall, Acta Polytech. Scand., Met. Ser., 1968, 74, 1 Search PubMed.
- Z. N. Wang, F. Liu, Y. A. Gao, W. C. Zhuang, L. M. Xu, B. X. Han, G. Z. Li and G. Y. Zhang, Langmuir, 2005, 21, 4931–4937 CrossRef CAS.
- T. Dürrschmidt and H. Hoffmann, Colloid Polym. Sci., 2001, 275, 1005–1015 Search PubMed.
- N. Granizo, M. Álvarez and M. Valiente, J. Colloid Interface Sci., 2006, 298, 363–368 CrossRef CAS.
- R. Mezzenga, C. Meyer, C. Servais, A. I. Romoscanu, L. Sagalowicz and R. C. Hayward, Langmuir, 2005, 21, 3322–3333 CrossRef CAS.
- S. M. Martin and M. D. Ward, Langmuir, 2005, 21, 5324–5331 CrossRef CAS.
- J. Li, M. W. Zhao and L. Q. Zheng, Colloids Surf., A, 2012, 396, 16–21 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra21467a |
| This journal is © The Royal Society of Chemistry 2012 |