Amorphous Si/SiOx/SiO2 nanocomposites via facile scalable synthesis as anode materials for Li-ion batteries with long cycling life†
Fang
Dai
,
Ran
Yi
,
Mikhail L.
Gordin
,
Shuru
Chen
and
Donghai
Wang
*
Department of Mechanical and Nuclear Engineering, The Pennsylvania State University, University Park, Pennsylvania 16802, United States. E-mail: dwang@psu.edu
First published on 23rd October 2012
Abstract
Novel Si/SiOx/SiO2 nanocomposites were prepared via a facile and scalable wet synthesis. The amorphous structure, nanoscale particle size, and composition of the Si/SiOx/SiO2 material were characterized and its electrochemical performance as an anode for Li-ion batteries was evaluated. The material shows a stable cycling capacity of ∼600 mA h g−1 over 350 cycles with high coulombic efficiency of >99%.
High-energy-density lithium ion batteries with long cycling lives have been regarded as promising energy storage devices for plug-in hybrid and electric vehicles.1 The development of high energy Li-ion batteries requires new anode materials with higher energy density than that of conventional graphite-based materials. Silicon has been considered as a promising high-energy anode because of its abundance and high theoretical capacity (>3500 mA h g−1).2 In spite of these advantages, the practical use of Si has thus far been hindered by significant capacity decay mainly due to the large volume changes (∼300%) and unstable solid electrolyte interphase (SEI) during the charging and discharging processes.3
Many attempts to improve the cycling stability of Si anodes have been made, such as designing porous structures,4 nanocrystalline particles,5 nanowires,6 hollow structures and core-shell structures.7 Besides metallic Si, Si alloys and Si-containing composites have also been investigated. Among those, SiOx-based composites are one of the most attractive candidates8 because of their excellent cycling performance.8j,9 During the lithiation process of SiOx, lithium oxide and lithium silicates are irreversibly formed and act as a buffer layer to tolerate the large volume change of the Si and facilitate formation of stable SEI layers.8f,h,9a,10 Previous studies also show that nano-sized SiOx materials are less susceptible to pulverization due to mechanical fracture by volume change during the charge and discharge processes, and thus have superior cycling stability to larger particles.11 For example, Chen et al. prepared an SiOx nanotube material which shows a reversible capacity of 940 mA h g−1.12 Park et al. reported a reversible capacity of 1280 mA h g−1 for a carbon coated Si/SiO/SiO2 composite.8k Yamamoto et al. showed a SiO/carbon nanofiber composite with a reversible capacity of 890 mA h g−1.13 However, only limited cycling performance (<100 cycles) has been reported for SiOx-based anode materials, except for the SiO/C composite reported by Osaka et al. (842 mA h g−1 after 7200 cycles).8l
To date, most synthesis routes of SiOx materials are through either oxidation of silicon or reduction of SiO2.8e,g,j Preparation of nano-sized SiOx in these manners is difficult because it requires nano-sized precursors that are themselves difficult to generate. This is especially true for the oxidation method, as preparation of the nano-sized Si precursor is usually performed by vapour deposition techniques and is thus hard to scale up, which has hindered the application of the corresponding SiOx materials in Li-ion batteries.14 In addition, carbon coating is indispensable for most SiOx materials in order to achieve good cycling performance, thus making the preparation process more complicated.8b,c,g–j
Here we report a new Si/SiOx/SiO2 anode material prepared via a facile and scalable route. Introducing SiCl4 as the Si source, the synthesis includes the formation of chloride-capped Si precursor by reduction of the SiCl4 in solution, followed by oxidation of the precursor. The characterization results suggest formation of amorphous Si/SiOx/SiO2 composite with nanoscale particle sizes. Without further surface or structural modification, the material shows an initial charge capacity of 610 mA h g−1, good cycling stability (maintained at >600 mA h g−1 after 350 cycles), and excellent coulombic efficiency after the initial cycles (average >99%).
The preparation of the Si/SiOx/SiO2 nanocomposite was carried out in a glovebox filled with Ar. The synthesis route (see ESI†) is shown in Scheme 1. Sodium potassium alloy (NaK) was slowly added to a toluene solution of SiCl4via a syringe. The reaction mixture was then heated under reflux for four hours. Fine black particles were formed during the reflux process. After treatment with diethyl ether solution of hydrogen chloride, the color of the particles changed to brown. These final products were removed from the glove box, washed with excess de-ionized water, and collected by filtration. The as-prepared material was annealed at 500 °C in Ar before use.
 | ||
| Scheme 1 Synthesis route of the Si/SiOx/SiO2 nanocomposite. | ||
Unlike other reducing reagents such as sodium naphthanide, which are used to obtain nanocrystalline Si,15 the NaK reducing reagent helps form an amorphous Si structure. The reduction of SiCl4 by NaK first forms chloride-terminated silicon particles. The terminal chloride ligands then react with H2O to form a terminal hydroxide structure (eqn (1)).15c Some hydrogen-terminated particles, which may be generated under the acidic conditions, also react with H2O to form terminal hydroxide structure (eqn (2)). H2O penetrates into the particle structure and the oxygen inserts into Si–Si bonds to form silicon oxide (eqn (3)). This Si oxidation process is similar to oxidation of a porous Si surface.16
| Si–Cl + H2O → Si–OH + HCl | (1) |
| Si–H + H2O → Si–OH + H2 | (2) |
| (Si–Si–)Si–OH + 2H2O → (Si–O–Si–O)Si–OH + 2H2 | (3) |
The oxygen content of the Si/SiOx/SiO2 nanocomposite was determined to be 42 wt% by thermogravimetric analysis (TGA) (Fig. S1†). Thus the material as a whole is identified as SiO1.3. The particle size and morphology were examined by transmission electron microscopy (TEM). The TEM images clearly show that the Si/SiOx/SiO2 takes the form of irregular aggregated particles with an average primary particle size of about 10 nm (Fig. 1A and Fig. S2†). In addition, no ordered lattice fringes are observed in the high-resolution TEM image, which shows the particles are amorphous (Fig. 1B). The X-ray powder diffraction (XRD) pattern of the nanocomposite after calcination is shown in Fig. 2A. It shows two broad peaks located at about 2θ = 28° and 50°, which are similar to those of SiO.8a,h,17 No obvious Si peaks are observed. Brunauer–Emmett–Teller (BET) surface area analysis indicates that the nanocomposite has a surface area of 257.5 m2 g−1. The isothermal sorption profile suggests that the material contains mesopores with Barrett–Joyner–Halenda (BJH) pore diameter of about 10 nm, which are the voids between aggregated nanoparticles (Fig. S3†). The energy-dispersive X-ray (EDS) spectrum confirms the presence of both silicon and oxygen (Fig. S4†). The X-ray photoelectron spectroscopy (XPS) of the nanocomposite shows a strong peak at about 103 eV with a broad shoulder, suggesting formation of SiOx and SiO2 (Fig. 2B).4a,5,7c,18 The other small peak at about 100 eV indicates a small amount of Si present in the particle.4a,5,7c,18 The appearance of the Si signal could be attributed to the Si remaining inside the particle without oxygen insertion. However, according to the HRTEM and XRD studies, even this remaining Si is amorphous (Fig. 1B). The integration of the peaks revealed that the ratio of metallic Si and silicon oxides was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5. It is impossible to give the exact ratio between SiOx and SiO2 due to the unknown x number of the SiOx.
:5. It is impossible to give the exact ratio between SiOx and SiO2 due to the unknown x number of the SiOx.
 | ||
| Fig. 1 (A) TEM image of Si/SiOx/SiO2 nanocomposite. (B) HRTEM image of the Si/SiOx/SiO2 nanoparticle in the composite. | ||
 | ||
| Fig. 2 Characterization of the Si/SiOx/SiO2 nanocomposite: (A) XRD pattern and (B) XPS spectrum. | ||
The structure of the Si/SiOx/SiO2 nanocomposite was further investigated by FT-IR spectroscopy (Fig. 3). The spectrum shows a peak at 1082 cm−1, which is assigned to the vibrational stretch of Si–O–Si belonging to silicon oxides (SiOx and SiO2).16c,19 The peak at 2260 cm−1 suggests the appearance of –Oy–SiHx, which arises from the oxidation of the surface silicon hydride structure. The broad peak around 3700 cm−1 is ascribed to –Si–OH vibrational stretch.16c,19 According to the XPS and FT-IR results, the particle structure could be described as a mixture of Si and silicon oxides which is terminated by oxide (–O), hydroxide (–OH), and hydride (–H) groups. The appearance of the vibrational stretches of Si–O–Si, –Oy–SiHx, and –Si–OH is evidence that the formation of the SiOx is due to the reaction of the functional group-terminated Si surface with H2O (eqn (3)). The appearance of the terminal oxide group (–O) suggests the formation of the SiO2 from the full oxidation of the Si.19 The exact ratio of SiOx and SiO2 is not clear yet due to the unknown number x of SiOx.
 | ||
| Fig. 3 FT-IR spectrum of the Si/SiOx/SiO2 nanocomposite. | ||
The electrochemical performance of the Si/SiOx/SiO2 material as an anode for Li-ion batteries was tested using CR2016 coin-type half-cells. The discharge–charge profiles of electrodes during the 1st, 2nd, 100th, and 350th cycles at 200 mA g−1 between 0.02 V and 1.5 V are shown in Fig. 4A. The first discharge capacity is 2190 mA h g−1, and the capacity stabilizes at ∼650 mA h g−1 for the cycles thereafter. The discharge–charge profiles show little change from the 2nd cycle to the 350th cycle. The low first cycle coulombic efficiency (∼30%) may be attributed to the formation of an SEI layer and irreversible formation of lithium oxides and lithium silicates within the nanocomposites.8j,9a,20
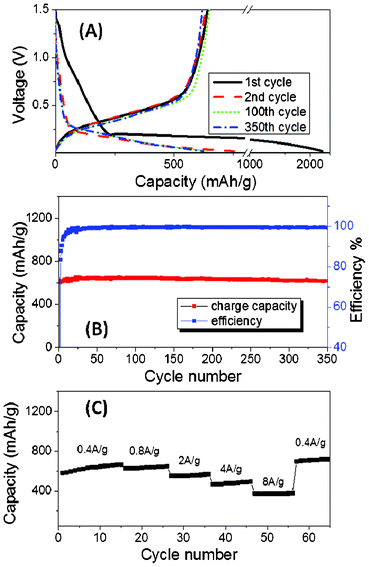 | ||
| Fig. 4 Electrochemical performance of the Si/SiOx/SiO2 nanocomposite. (A) Discharge–charge profiles obtained after 1st, 2nd, 100th and 350th cycles. (B) Charge capacity and coulombic efficiency of 350 cycles between 0.02–1.5 V. (C) Charge capacity at various rates from 0.4 A g−1 to 8 A g−1. | ||
The nanocomposite shows good cycling stability. The cycling performance at 200 mA g−1 between 0.02 V and 1.5 V is shown in Fig. 4B. After the initial cycle, the capacity increases until it peaks at 650 mA h g−1 after about 30 cycles. This increase is attributed to the slow activation of the Si/SiOx-containing material. The material then shows good capacity retention after 100 cycles (>99%) and after 350 cycles (>95%) compared with the peak capacity. The coulombic efficiencies reach 99.5% after around 30 cycles and remain at >99.5%.
The rate performance of the nanocomposite anode was also tested at current densities of 0.4 A g−1, 0.8 A g−1, 2 A g−1, 4 A g−1, and 8 A g−1. The results are shown in Fig. 4C. The anode shows good coulombic efficiency (>99%) even at a current density of 8 A g−1. The capacity at this current density is still 375 mA h g−1, which is comparable to the theoretical capacity of graphite. A capacity of >630 mA h g−1 is again observed when the current density is reset to 0.4 mA g−1 after 60 cycles, showing that the electrode was not damaged by the high rate test.
The excellent cycling stability of the Si/SiOx/SiO2 nanocomposite is attributed to its nanoscale particle size and the amorphous SiOx-containing structure. The small particle size leads to higher surface area and thus better electrical contact with the conductive additive than is the case for larger particles. In addition, fracture can be avoided because the tension generated in nanoparticles can be easily relaxed due to their small size.11 The silicon oxides also act as buffer layers for the large volume change of the Si during the discharging-charging process.8f,8h,9a,10a,b Finally, the amorphous structure of the composite could prevent pulverization due to the isotropic expansion during lithium insertion, as compared to the highly anisotropic expansion of crystalline Si. The capacity loss of the material is mainly due to the irreversible formation of lithium oxides and lithium silicates during lithiation of the SiOx.8j,9a,20 However, although the use of SiOx rather than metallic Si requires sacrificing some capacity, this formation of buffer layers grants the material excellent volume change tolerance.8e,8g,8l,9b,10a
In conclusion, we synthesized a novel amorphous Si/SiOx/SiO2 composite with nanoscale particle size. The solution-based synthesis under mild reaction conditions is easy to scale up. Electrochemical characterization reveals that the material has a very stable capacity and solid rate performance. The silicon oxide-containing amorphous structure of the Si/SiOx/SiO2 nanocomposite can effectively accommodate the volume change of Si during the discharging–charging process. The relationship between the content of silicon oxides in the composite and the anode performance, such as the first cycle coulombic efficiency and cycling stability, is under further investigation.
This work was supported by the Assistant Secretary for Energy Efficiency and Renewable Energy, Office of Vehicle Technologies of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231, Subcontract NO. 6951378 under the Batteries for Advanced Transportation Technologies (BATT) Program.
References
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS.
- B. A. Boukamp, G. C. Lesh and R. A. Huggins, J. Electrochem. Soc., 1981, 128, 725–729 CrossRef CAS.
- (a) K. Xu, Chem. Rev., 2004, 104, 4303–4418 CrossRef CAS; (b) C. K. Chan, R. Ruffo, S. S. Hong and Y. Cui, J. Power Sources, 2009, 189, 1132–1140 CrossRef CAS.
- (a) H. Kim, B. Han, J. Choo and J. Cho, Angew. Chem., Int. Ed., 2008, 47, 10151–10154 CrossRef CAS; (b) J. C. Guo, A. Sun and C. S. Wang, Electrochem. Commun., 2010, 12, 981–984 CrossRef CAS; (c) A. Magasinski, P. Dixon, B. Hertzberg, A. Kvit, J. Ayala and G. Yushin, Nat. Mater., 2010, 9, 353–358 CrossRef CAS.
- H. Kim, M. Seo, M.-H. Park and J. Cho, Angew. Chem., Int. Ed., 2010, 49, 2146–2149 CrossRef CAS.
- (a) C. K. Chan, H. Peng, G. Liu, K. McIlwrath, X. F. Zhang, R. A. Huggins and Y. Cui, Nat. Nanotechnol., 2008, 3, 31–35 CrossRef CAS; (b) J. W. Choi, J. McDonough, S. Jeong, J. S. Yoo, C. K. Chan and Y. Cui, Nano Lett., 2010, 10, 1409–1413 CrossRef CAS.
- (a) S. Chen, M. L. Gordin, R. Yi, G. Howlett, H. Sohn and D. Wang, Phys. Chem. Chem. Phys., 2012, 14, 12741–12745 RSC; (b) X. L. Li, P. Meduri, X. L. Chen, W. Qi, M. H. Engelhard, W. Xu, F. Ding, J. Xiao, W. Wang, C. M. Wang, J. G. Zhang and J. Liu, J. Mater. Chem., 2012, 22, 11014–11017 RSC; (c) N. Liu, H. Wu, M. T. McDowell, Y. Yao, C. Wang and Y. Cui, Nano Lett., 2012, 12, 3315–3321 CrossRef CAS.
- (a) J. Yang, Y. Takeda, N. Imanishi, C. Capiglia, J. Y. Xie and O. Yamamoto, Solid State Ionics, 2002, 152–153, 125–129 CrossRef CAS; (b) H.-Y. Lee and S.-M. Lee, Electrochem. Commun., 2004, 6, 465–469 CrossRef CAS; (c) T. Morita and N. Takami, J. Electrochem. Soc., 2006, 153, A425–A430 CrossRef CAS; (d) J. H. Kim, H. J. Sohn, H. Kim, G. Jeong and W. Choi, J. Power Sources, 2007, 170, 456–459 CrossRef CAS; (e) T. Zhang, J. Gao, H. P. Zhang, L. C. Yang, Y. P. Wu and H. Q. Wu, Electrochem. Commun., 2007, 9, 886–890 CrossRef CAS; (f) C. H. Doh, C. W. Park, H. M. Shin, D. H. Kim, Y. D. Chung, S. I. Moon, B. S. Jin, H. S. Kim and A. Veluchamy, J. Power Sources, 2008, 179, 367–370 CrossRef CAS; (g) Y.-S. Hu, R. Demir-Cakan, M.-M. Titirici, J.-O. Müller, R. Schlögl, M. Antonietti and J. Maier, Angew. Chem., Int. Ed., 2008, 47, 1645–1649 CrossRef CAS; (h) A. Veluchamy, C. H. Doh, D. H. Kim, J. H. Lee, D. J. Lee, Y. H. Ha, H. M. Shin, B. S. Jin, H. S. Kim, S. I. Moon and C. W. Park, J. Power Sources, 2009, 188, 574–577 CrossRef CAS; (i) A. Guerfi, P. Charest, M. Dontigny, J. Trottier, M. Lagacé, P. Hovington, A. Vijh and K. Zaghib, J. Power Sources, 2011, 196, 5667–5673 CrossRef CAS; (j) J. Wang, H. L. Zhao, J. C. He, C. M. Wang and J. Wang, J. Power Sources, 2011, 196, 4811–4815 CrossRef CAS; (k) J. I. Lee, N. S. Choi and S. Park, Energy Environ. Sci., 2012, 5, 7878–7882 RSC; (l) H. Nara, T. Yokoshima, T. Momma and T. Osaka, Energy Environ. Sci., 2012, 5, 6500–6505 RSC.
- (a) H. Yamamura, K. Nobuhara, S. Nakanishi, H. Iba and S. Okada, J. Ceram. Soc. Jpn., 2011, 119, 855–860 CrossRef CAS; (b) J. H. Kim, C. M. Park, H. Kim, Y. J. Kim and H. J. Sohn, J. Electroanal. Chem., 2011, 661, 245–249 CrossRef CAS.
- (a) J.-I. Lee, N.-S. Choi and S. Park, Energy Environ. Sci., 2012, 5, 7878 RSC; (b) Y. Idota, Science, 1997, 276, 1395–1397 CrossRef CAS.
- (a) J. Yang, M. Winter and J. O. Besenhard, Solid State Ionics, 1996, 90, 281–287 CrossRef CAS; (b) K. Zhao, M. Pharr, J. J. Vlassak and Z. Suo, J. Appl. Phys., 2011, 109, 016110–016113 CrossRef.
- H. Guo, R. Mao, X. Yang and J. Chen, Electrochim. Acta, 2012, 74, 271–274 CrossRef CAS.
- Q. Si, K. Hanai, T. Ichikawa, M. B. Phillipps, A. Hirano, N. Imanishi, O. Yamamoto and Y. Takeda, J. Power Sources, 2011, 196, 9774–9779 CrossRef CAS.
- P. R. Abel, Y. M. Lin, H. Celio, A. Heller and C. B. Mullins, ACS Nano, 2012, 6, 2506–2516 CrossRef CAS.
- (a) R. A. Bley and S. M. Kauzlarich, J. Am. Chem. Soc., 1996, 118, 12461–12462 CrossRef CAS; (b) D. Mayeri, B. L. Phillips, M. P. Augustine and S. M. Kauzlarich, Chem. Mater., 2001, 13, 765–770 CrossRef CAS; (c) J. Zou, P. Sanelle, K. A. Pettigrew and S. M. Kauzlarich, J. Cluster Sci., 2006, 17, 565–578 CrossRef CAS.
- (a) T. Unagami, Jpn. J. Appl. Phys., 1980, 19, 231–241 CrossRef CAS; (b) V. Chirvony, A. Chyrvonaya, J. Ovejero, E. Matveeva, B. Goller, D. Kovalev, A. Huygens and P. de Witte, Adv. Mater., 2007, 19, 2967 CrossRef CAS; (c) D. B. Mawhinney, J. A. Glass and J. T. Yates, J. Phys. Chem. B, 1997, 101, 1202–1206 CrossRef CAS.
- C. M. Park, W. Choi, Y. Hwa, J. H. Kim, G. Jeong and H. J. Sohn, J. Mater. Chem., 2010, 20, 4854–4860 RSC.
- A. A. Balandin and K. L. Wang, Handbook Of Semiconductor Nanostructures And Nanodevices, American Scientific Publishers, 2005 Search PubMed.
- M. Niwano, J.-I. Kageyama, K. Kinashi, J.-I. Sawahata and N. Miyamoto, Surf. Sci., 1994, 301, L245–L249 CrossRef CAS.
- (a) B. K. Guo, J. Shu, Z. X. Wang, H. Yang, L. H. Shi, Y. N. Liu and L. Q. Chen, Electrochem. Commun., 2008, 10, 1876–1878 CrossRef CAS; (b) X. L. Yang, Z. Y. Wen, L. Zhang and M. You, J. Alloys Compd., 2008, 464, 265–269 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ra22187j |
| This journal is © The Royal Society of Chemistry 2012 |