Free-lignin cellulose obtained from agar industry residues using a continuous and minimal solvent reaction/extraction methodology
R.
López-Simeon
ac,
J.
Campos-Terán
c,
H. I.
Beltrán
*b and
M.
Hernández-Guerrero
*c
aUniversidad Autónoma Metropolitana, Unidad Iztapalapa, San Rafael Atlixco 186 Col. Vicentina, C.P. 09340, México, D.F., México
bUniversidad Autónoma Metropolitana, Unidad Cuajimalpa, Departamento de Ciencias Naturales, Pedro Antonio de los Santos 84, Col. San Miguel Chapultepec, 11850, D.F, México. E-mail: hbeltran@correo.cua.uam.mx; Tel: (55) 2636 3806
cUniversidad Autónoma Metropolitana Unidad Cuajimalpa, Departamento de Procesos y Tecnología, Pedro Antonio de los Santos 84, Col. San Miguel Chapultepec, 11850, D.F, México. E-mail: mhernandez@correo.cua.uam.mx; Tel: (55) 2636 3818
First published on 25th September 2012
Abstract
Herein we present a green procedure to obtain cellulose (Cel) polymers with different physicochemical properties at high purity, and underline its two major aspects: sustainability and efficiency. In the first place, regarding sustainability, the source of Cel was residues from agar industries, which are based on red seaweed and hence free of lignin, thus facilitating the extraction of Cel. In the aspect of efficiency, a continuous extraction/reaction system was used to obtain pure Cel from these residues. The extraction/reaction device used in this study normally works in a liquid–liquid extraction fashion, but in this particular case it was successfully employed as a liquid–solid system. This methodology is important, because it concomitantly reduces the time of extraction/reaction procedures in the same flask and also minimizes the amount of solvent used. Thus high purity Cel was obtained using a continuous and minimal solvent extraction/reaction system in neutral/acidic/basic conditions leading to Celn/Cela/Celb polymers in 42/34/43.3% yield. These materials were characterized by 13C cross-polarization magic-angle spinning (CP-MAS) NMR, Fourier transform infrared spectroscopy (FT-IR), CHNS elemental analyses, X-ray diffraction (XRD), size exclusion chromatography (SEC) and compared against microcrystalline cellulose (MCC), confirming chemical integrity. Crystallinity index (CI [%]), was obtained from XRD/CP-MAS NMR data. All samples had slightly higher crystallinity than that of MCC. Molecular weight (MW, g mol−1), polydispersity index (PDI) and degree of polymerization (DP) for Celn, Cela, Celb polymers were all higher than those in MCC. Compared to MCC, the physicochemical characteristics of the isolated Cel polymers varied depending on the treatment, neutral being the mildest. The greener procedures developed herein provide Cel suitable for research and development of Cel derived substances.
Introduction
Cellulose (Cel) is the main structural component of plants and algae, as well as one of the most abundant and renewable biomolecules.1,2 It is a widely used polymer in the chemical, pharmaceutics and fuel industries, but its extraction from higher plants is limited by the presence of lignin, which is a very recalcitrant biopolymer.3 In contrast to higher plants, some algae,4 including red algae (Rhodophyta), do not contain lignin hence Cel extraction from algal resources is a simple alternative to the methods of extraction from plants. Red algae belong to the Protist kingdom, superphylum Algae and phylum Rhodophyta. Most red algae are multicellular, autotrophic and photosynthetic seaweeds. They have a double cell wall, their external wall being formed mainly of long chain polysaccharides, predominantly Cel, while the internal wall is constituted by xylans, mannans and sulfonated galactans.5–7 Rhodophyta also contain phycoerythrin, a red protein, which is responsible for their characteristic red tint. This phycoerythrin masks the green color from the chlorophyll in these algae.8 Rhodophyta are easily cultivated in situ and are therefore widely used in industry.5,9,10 Some industrial products from these algae include antibiotics, fertilizers, agglutinants, forage, laxatives, clarifying agents and saccharides, such as glucose.11 However, Rhodophyta are mainly used in the food industry for the extraction of hydrocolloids, also called phycolloids.6 The most important phycocolloids are agar and carrageenans. Agar and carrageenans are natural high molecular weight polysaccharides that provide the structural support in the cell walls of the algae. The two main Rhodophyta used in the agar industry are Gelidium sp. and Gracilaria sp., other minor algae include Pterocladia sp., Gelidiella sp., Aconthopeltis sp. and Annfeltia sp..12–14 In general, these algae are called agarophytic. As of 2009, the annual world production of agar was 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 ton.15 In Mexico, the annual production of agarophytic algae is estimated as 5153 ton (hydrated algae) per year.16 The agar production is calculated to be about the 25% (dry weight) of the algae and represents approximately 582 tons per year.17 The main species used for agar and carrageenan production in Mexico are Gelidium robustum, Gracilariopsis lameniformis and Chondracanthus canaliculatus.18,19 Agar is formed by agarose and agaropectin, the proportion of each fraction depends on the origin of the algae. Agar is an excellent viscosity modifier, gelling agent, emulsifier and stabilizer.11 Apart from its use in the food industry, the agarose fraction of agar is also used for separations. The agarose matrix (a linear polymer) is considered an ideal medium for polymer diffusion, as it is a neutral polysaccharide and is therefore adequate for gel electrophoresis and chromatography. Agar is also a very important product in bacteriology, where it is used as a microbiological culture media due to its gelling thermal reversibility. All methods used for agar extraction are based on common methodologies, including manual cleaning, drying and boiling in water for at least three cycles. This is followed by an acidic treatment at pH 4 with either 1% H2SO4 or 10% acetic acid. The samples are then filtered and finely ground.7,14,20,21 It is worthwhile mentioning that the algae does not degrade completely during agar extraction. The extraction of the phycocolloid mainly affects the internal wall leaving behind the external (Cel-rich) wall as residue. Consequently, it is possible to process the low-value residues of the agar industry as a source of Cel, opening the possibility of obtaining products of added-value.22,23Cel extraction in seaweeds is commonly done in vials or a Soxhlet equipment, with large amounts of solvents and over long periods of time.24,25 However, there is a more convenient system, which is commonly used in liquid–liquid extractions for the recovery of organic traces, mainly contaminants or pesticides. This system achieves continuous solution enrichment and consequently an optimal extraction of different products.26,27 Additionally, this system has other advantages: the use of solvent is minimized, it has a recovery of about 85% or even more, and the time of extraction is reduced.28,29 For all these reasons this is an environmentally friendly option and is also a low energy consuming method. This type of system was only used in liquid–liquid preconcentration/extraction, but in this work we have successfully used it in a solid–liquid system for the extraction of all organic and inorganic non Cel related trace compounds in agar residues. The extraction of Cel from these residues was achieved under neutral, acidic or basic conditions and it was facilitated due to the absence of lignin and other polymers of different molecular weights. The extracted Cel samples were characterized by FT-IR, 13C CP-MAS NMR, CHNS elemental analyses, XRD and were compared to a commercial MCC. The CI was obtained from XRD and NMR data. Properties such as crystallite sizes, molecular weights and polydispersity indices for each of the extracted samples are also reported.
500 ton.15 In Mexico, the annual production of agarophytic algae is estimated as 5153 ton (hydrated algae) per year.16 The agar production is calculated to be about the 25% (dry weight) of the algae and represents approximately 582 tons per year.17 The main species used for agar and carrageenan production in Mexico are Gelidium robustum, Gracilariopsis lameniformis and Chondracanthus canaliculatus.18,19 Agar is formed by agarose and agaropectin, the proportion of each fraction depends on the origin of the algae. Agar is an excellent viscosity modifier, gelling agent, emulsifier and stabilizer.11 Apart from its use in the food industry, the agarose fraction of agar is also used for separations. The agarose matrix (a linear polymer) is considered an ideal medium for polymer diffusion, as it is a neutral polysaccharide and is therefore adequate for gel electrophoresis and chromatography. Agar is also a very important product in bacteriology, where it is used as a microbiological culture media due to its gelling thermal reversibility. All methods used for agar extraction are based on common methodologies, including manual cleaning, drying and boiling in water for at least three cycles. This is followed by an acidic treatment at pH 4 with either 1% H2SO4 or 10% acetic acid. The samples are then filtered and finely ground.7,14,20,21 It is worthwhile mentioning that the algae does not degrade completely during agar extraction. The extraction of the phycocolloid mainly affects the internal wall leaving behind the external (Cel-rich) wall as residue. Consequently, it is possible to process the low-value residues of the agar industry as a source of Cel, opening the possibility of obtaining products of added-value.22,23Cel extraction in seaweeds is commonly done in vials or a Soxhlet equipment, with large amounts of solvents and over long periods of time.24,25 However, there is a more convenient system, which is commonly used in liquid–liquid extractions for the recovery of organic traces, mainly contaminants or pesticides. This system achieves continuous solution enrichment and consequently an optimal extraction of different products.26,27 Additionally, this system has other advantages: the use of solvent is minimized, it has a recovery of about 85% or even more, and the time of extraction is reduced.28,29 For all these reasons this is an environmentally friendly option and is also a low energy consuming method. This type of system was only used in liquid–liquid preconcentration/extraction, but in this work we have successfully used it in a solid–liquid system for the extraction of all organic and inorganic non Cel related trace compounds in agar residues. The extraction of Cel from these residues was achieved under neutral, acidic or basic conditions and it was facilitated due to the absence of lignin and other polymers of different molecular weights. The extracted Cel samples were characterized by FT-IR, 13C CP-MAS NMR, CHNS elemental analyses, XRD and were compared to a commercial MCC. The CI was obtained from XRD and NMR data. Properties such as crystallite sizes, molecular weights and polydispersity indices for each of the extracted samples are also reported.
Experimental section
The residues from agar industries and seaweed were kindly donated by Agarmex (Ensenada, Baja California, Mexico). Ultra pure water with 18 mΩ resistivity was obtained from a water purifying system (Milli-Q synthesis, Millipore, France). Unless otherwise specified, all reagents listed below were purchased from Sigma-Aldrich (Milwaukee, WI, USA). MCC of high purity with a particle size of ca. 20 μm (powder), methanol (MeOH, reagent grade ≥99.8%), hydrochloric acid (HCl, reagent grade, assay 37%), sodium hydroxide (NaOH, reagent grade, assay 80%), sodium chlorite (NaClO2, technical grade, assay 80%), potassium bromide (KBr, reagent grade, ≥99.0%), lithium chloride (LiCl, anhydrous, ≥99.9%), N,N-dimethylacetamide (DMAc, Chromasolv plus-HPLC, ≥99.9%). The acetate solution (pH 5) was prepared by mixing 357 mL (0.2 M acetic acid, reagent grade ≥99.7%) and 643 mL (0.2 M sodium acetate) of free and metallic acetates respectively. Vanadium pentoxide (≥99.9%) was purchased and used as received from Thermo Scientific (Waltham, MA, USA).Residues cleaning
The residues from the phycocolloid extraction have some impurities such as sand, salts and calcareous deposits in the cell wall, which form part of the exoskeleton of the algae, shells and other organisms. The residues also contain sulfolipids, phycobiliproteins (pigments), nucleic acids, D-xilose and Cel.30 Therefore a pre-extraction conditioning was necessary to ensure homogeneity in the samples. The sample conditioning consisted of several steps, including manual cleaning, salt elimination by washing with ultrapure water, drying, grinding and sieving. A detailed description of each step is given below.Manual cleaning
The manual cleaning of the samples consisted of eliminating the remaining calcareous material, shells and other organisms prior to the next step. Sieving was also necessary to eliminate some salts and sand.Salt elimination
The residues were washed at room temperature to eliminate the remaining sand, salts and chemicals used during the agar extraction. For the washing, 100 g of sample were suspended in 1 L of ultrapure water and shaken. Water was exchanged every 24 h over two days. After this period of time, it was exchanged every 72 h. Important variables such as pH, conductivity and total dissolved solids were measured before each water exchange until the final readings of pH and conductivity were those for ultrapure water. The complete washing of the residues was achieved after 250 h. Once the samples were cleaned, the excess of water was removed and the solids were dried in an oven at 65 °C until they were at constant weight. The dried samples were ground, strained through a 70 mesh sieve and stored under dry conditions.The isolation of Cel from the agar industry residues was accomplished in a very similar fashion to the extraction of Cel directly from algae.24,31,32 However, some chemical and equipment modifications were made. This new methodology reduces both the time of extraction/reaction in the same flask and the amount of solvent used for it. Two steps of the general treatment described below were varied, namely the methanolic wash and the bleaching, thus modifying the nature of the extraction solvent using acidic, neutral or alkaline conditions. The general extraction procedure consisted firstly of a methanolic wash (in neutral, acidic or basic conditions as mentioned above), then bleaching followed by acidic and basic washings, and finally neutralization and drying. A detailed description of each stage is provided below. A continuous extraction system was used and, in every step (Scheme 1), the reaction/extraction time was shorter compared to the same procedure done in normal reaction vials. Furthermore, the volume of solvents and chemicals was minimized, as these were recycled and could also be recovered.
 | ||
| Scheme 1 Continuous extraction/reaction apparatus. Left, the extraction system: extractor body (Flask 1) a1) solvent and solid deposit, a2) evaporator connector, a3) solvent return line with Teflon valve, b) condenser, c) collecting round bottom flask (Flask 2). Right, solvent deposit with pure Cel after treatment. | ||
The extraction system contained a liquid–liquid reaction/extraction glass apparatus with an extractor body (Scheme 1, a1) consisting of a long necked oval flask with two side-arms, a condenser connected from the neck of the extractor body (Scheme 1, b) and a collecting round bottom flask (Scheme 1, c). The evaporator connector, a 90° side-arm (Scheme 1, a2) located at the middle of the neck of the extraction body, is attached to the collecting flask. As was already mentioned above, the continuous extraction/reaction device that was used in this work is of a liquid–liquid nature, but in this particular case it was used for a liquid–solid system. It was designed to provide a continuous extraction of the non-cellulose contaminants in agar residues, which was achieved with free distilled solvent (due to its evaporation and condensation from the collecting flask to the extractor body that contained the residues, see Scheme 1, a1). The system is connected between flasks with a liquid decanter and a Teflon® valve with a cotton filter allowing the flow of soluble species (Scheme 1, a3).
Methanolic wash
During this step either an acidic (HCl) or a basic (NaOH) reagent was added together with methanol, resulting in three different extraction procedures. Cellulose samples (Celn, Cela and Celb) with different characteristics were therefore extracted depending on the nature of the methanolic wash. Celn was obtained in pure MeOH, Cela in the MeOH + HCl extraction/reaction system and Celb in the MeOH + NaOH system. A detailed description of each system is given below. The sieved and clean residue sample (5 g) was placed in the solvent extraction body (see Scheme 1, a1) together with the solvent (MeOH, 50 mL) or solvent mixture, either MeOH + 10 μL HCl per g of sample or MeOH + 0.04 g NaOH per g of sample. The collecting flask (Scheme 1, c) was also filled with solvent or MeOH mixture (50 mL). The contents of the collecting flask were heated to maintain reflux, thus ensuring a continuous solvent exchange with the extraction body. Optimal removal of methanol soluble contaminants in the sample was reached after four days.Bleaching
The methanol/methanol mixture was removed and the sample was bleached by adding 50 mL of acetate solution (pH 5) and 2.5 g of NaClO2 to the extraction body. The sample was then heated at 30 °C for 12 h. Subsequently, the samples were neutralized with ultrapure water under reflux in the reaction/extraction apparatus over 48 h. The aqueous solution was changed to ultrapure water every 24 h.Basic washing
After neutralization, the samples were subjected to a basic washing in which 50 mL of an aqueous NaOH solution (0.5 M) per g of sample were added. The samples were heated to 60 °C over 12 h and neutralized following the same neutralization procedure described in the bleaching step.Acidic washing
The last step for the extraction was an acidic wash. 35 mL of an aqueous solution of HCl (5% v/v) per g of sample were added. The sample was heated to boiling and, immediately after this, the samples were neutralized over 72 h with water changes every 24 h. Finally, the extracted Cel was dried in an oven at 65 °C. Yields were calculated, both on the basis of initial amount of agar residues (mg extracted Cel per g residue) and taking into consideration the amount of glucose (58.1%) in red algae Gelidium after agar extraction as determined by Vigon, et al. in 1994.33Characterization
FT-IR
Celn, Cela and Celb were characterized by FT-IR (Pressurized ATR accessory, Bruker Tensor 27 FT-IR spectrophotometer, USA; the spectra were acquired after 1024 scans). The characteristic IR absorption bands corresponding to O–H, C–H and C–O chemical bonds present in the extracted polymers were used to assess their chemical nature. Furthermore, the FT-IR spectrum of MCC was used as standard for comparison purposes.13C CP-MAS NMR
Common pulse sequence from Bruker was employed for the acquisition of 13C CP-MAS NMR spectra of Celn, Cela, Celb and MCC samples34 (ASX300 Bruker, 300 MHz and CP-MAS probe) at 75.435 MHz.CHNS analyses
The elemental analyses of C, H, C/N and C/S were determined using a CHNS/O analyzer Flash 2000 series from Thermoscientific. Celn, Cela, Celb and MCC samples were weighed out ranging from 2.5 to 3 mg ± 0.5 mg in tin capsules and vanadium pentoxide (10 mg ± 0.5 mg) was added as a desiccant. The chromatograms were calibrated against a methionine standard and the results were obtained as calculated in percent of each element by the Eager 300 analysis software.XRD
The XRD patterns of the Celn, Cela, Celb and MCC samples were measured in air at room temperature with a Bruker AXS™ D-8 Advance diffractometer with the Bragg–Brentano 0-0 geometry, Cu-Kα radiation, a Ni 0.5% CuKβ filter in the secondary beam, and a 1-dimensional position sensitive silicon strip detector (Bruker, Linxeye). The diffraction intensity as a function of the 2θ angle was measured between 6.5° and 110°, with a 2θ step of 0.039° for 52.8 s per point.Cristallinity indices (CI)
The CI of the Celn, Cela, Celb and MCC samples was determined through XRD and 13C CP-MAS NMR data analysis. In the XRD diffractograms, two different methods were used to obtain the CI. First, the CIXRD-PHR (cristallinity index by XRD-peak high ratio) was calculated from the height ratio between the intensity of the peak (I[002]-I[AM]), and total intensity (I[002]) after subtraction of the background signal, where I002 is the point with the highest intensity in the [002] peak, and IAM is the lowest value of the depression between the [10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ] and [002] peaks.35 In the second method, the areas of the crystalline and amorphous parts were determined through integration and the CIXRD (cristallinity index by XRD) obtained as the ratio of the crystalline area to the total area (both amorphous and crystalline regions). In the NMR spectra, two signals of the C4 carbon, at 89 ppm and at 84 ppm, characteristic of the crystalline and amorphous fractions of the sample were used as reference to calculate CINMR. The area of the crystalline peak (from 87 to 93 ppm) was divided by the total area belonging to both crystalline and amorphous C4 signal contributions (from 80 to 93 ppm).36
] and [002] peaks.35 In the second method, the areas of the crystalline and amorphous parts were determined through integration and the CIXRD (cristallinity index by XRD) obtained as the ratio of the crystalline area to the total area (both amorphous and crystalline regions). In the NMR spectra, two signals of the C4 carbon, at 89 ppm and at 84 ppm, characteristic of the crystalline and amorphous fractions of the sample were used as reference to calculate CINMR. The area of the crystalline peak (from 87 to 93 ppm) was divided by the total area belonging to both crystalline and amorphous C4 signal contributions (from 80 to 93 ppm).36
Crystallite average size (dp)
The dp of the Celn, Cela, Celb and MCC samples was calculated from the Scherrer equation: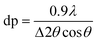 | (1) |
SEC
Molecular weights and molecular weight distributions were determined by SEC on an Agilent 1200 Series Modular Liquid Chromatography (LC) system comprised of a quaternary pump, a standard autosampler, a thermostated column compartment and UV-Vis and differential refractive index detectors. The stationary phase consisted of two PL-Gel 10 μm (7.5 mm id) Mixed-B columns. Solutions for chromatography were prepared with Cel:LiCl:DMAc (6.0:8.5:85.5),1,38 stirred for 72 h and filtered with 0.45μm polytetrafluoroethylene (PTFE) membranes. DMAc with 8% w/v LiCl was used as eluent at a flow rate of 1 mL min−1 at 40 °C. The system was calibrated with commercial narrow (Polymer Laboratories) pullulan standards, molecular weight range 180 to 780000 Da.39 SEC traces were analyzed to obtain 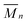 ,
, 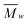 and PDI (
and PDI (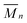 /
/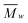 ) with the Polymer Labs Cirrus GPC/SEC Software Version 3.1.
) with the Polymer Labs Cirrus GPC/SEC Software Version 3.1.
Degree of polymerization (DP)
The DP was calculated taking anhydroglucose as the repeating unit, and dividing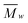 with this monomeric molecular fragment weight.
with this monomeric molecular fragment weight.
Characterization and spectroscopic data
Agar residue
IR (ν, cm−1, ATR): 3652–2994 (O–H), 2927–2854 (C–H),1573–1325 (C–OCO3), 1163 (C–O–C), 1056 (C–O), 1028 (C–O), 871 (C–OCO3), 713 (C–OCO3), 663. 13C NMR (δ, ppm): 177.1, 172.2, 168.3 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Oproteins), 136.0, 129.2, 127.2 (Caromatic[proteins]) 105.0, 103.6, 101.2 (C1), 88.5 (C4cryst), 83.5 (C4amorph), 74.4, 72.0 (C2, 3, 5), 64.7 (C6cryst), 61.8 (C6amorph), 40.3, 30.4, 21.2 (Caliphatic[proteins]). XRD (°, 2θ (count number) [h k l]): 48.8 (1539)[016], 47.5 (1635)[018], 43.3 (1779)[202], 39.5 (2115)[113], 36.1 (2019)[010], 29.5 (6490)[104], 28.2 (3365)[012], 26.7 (5962)[111], 23.0 (3894)[002], 15.5 (2596)[101] [10]. Elemental analysis, experimental (%): C, 61.66; H, 8.51, N, 6.09, S, 0.27.
Oproteins), 136.0, 129.2, 127.2 (Caromatic[proteins]) 105.0, 103.6, 101.2 (C1), 88.5 (C4cryst), 83.5 (C4amorph), 74.4, 72.0 (C2, 3, 5), 64.7 (C6cryst), 61.8 (C6amorph), 40.3, 30.4, 21.2 (Caliphatic[proteins]). XRD (°, 2θ (count number) [h k l]): 48.8 (1539)[016], 47.5 (1635)[018], 43.3 (1779)[202], 39.5 (2115)[113], 36.1 (2019)[010], 29.5 (6490)[104], 28.2 (3365)[012], 26.7 (5962)[111], 23.0 (3894)[002], 15.5 (2596)[101] [10]. Elemental analysis, experimental (%): C, 61.66; H, 8.51, N, 6.09, S, 0.27.
Celn
White solid, yield (%) 42 (mg Celn per g residue) (72.4 glucose based); IR (ν, cm−1, ATR): 3615–2984 (O–H), 2926–2843 (C–H), 1433, 1308, 1157 (C–O–C), 1101 (C–O), 1056 (C–O), 1028 (C–O), 898 (O–Canom–H), 662. 13C NMR (δ, ppm): 104.8 (C1), 88.2 (C4cryst), 83.3 (C4amorph), 74.2, 71.9 (C2, 3, 5), 64.6 (C6cryst), 62.0 (C6amorph). XRD (°, 2θ (count number) [h k l]): 34.4 (1803)[040], 22.6 (7985)[002], 15.6 (3734)[101] [10]; dp (Å): 40.1[002]; 38.0[040]. CI (%): 72.8XRD-PHR; 47.4XRD; 55.7NMR. SEC (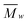 [g mol−1], PDI, DP): 148528 (11.82) 917. Elemental analysis, experimental (%): C, 41.42; H, 5.93. Calculated: for anhydroglucose (C6H10O5): C, 44.45; H, 6.22; and for glucose (C6H12O6): C, 40.00; H, 6.71.
[g mol−1], PDI, DP): 148528 (11.82) 917. Elemental analysis, experimental (%): C, 41.42; H, 5.93. Calculated: for anhydroglucose (C6H10O5): C, 44.45; H, 6.22; and for glucose (C6H12O6): C, 40.00; H, 6.71.
Cela
White solid, yield (%) 34 (mg Cela per g residue) (58.6 glucose based); IR (ν, cm−1, ATR): 3589–2961 (O–H), 2926–2858 (C–H), 1430, 1313, 1163 (C–O–C), 1108 (C–O), 1060 (C–O), 1026 (C–O), 889 (O–Canom–H), 664. 13C NMR (δ, ppm): 105.2–104.1 (C1), 88.5 (C4cryst), 84.0–83.1 (C4amorph), 74.5, 71.9, 71.2 (C2, 3, 5), 64.7 (C6cryst), 62.2 (C6amorph). XRD (°, 2θ (count number)[h k l]): 34.3 (2285)[040], 22.5 (12395)[002], 15.4 (5763)[101][10]; dp (Å): 45.1[002]; 53.1[040]. CI (%): 72.6XRD-PHR; 51.2XRD; 59.3NMR. SEC (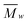 [g mol−1], PDI, DP: 78857 (7.22) 487. Elemental analysis, experimental (%): C, 41.27; H, 5.93. Calculated: for anhydroglucose (C6H10O5): C, 44.45; H, 6.22; and for glucose (C6H12O6): C, 40.00; H, 6.71.
[g mol−1], PDI, DP: 78857 (7.22) 487. Elemental analysis, experimental (%): C, 41.27; H, 5.93. Calculated: for anhydroglucose (C6H10O5): C, 44.45; H, 6.22; and for glucose (C6H12O6): C, 40.00; H, 6.71.
Celb
White solid, yield (%) 43 (mg Celb/g residue) (74.6 glucose based); IR (ν, cm−1, ATR): 3727–2977 (O–H), 2930–2852 (C–H), 1430, 1313, 1156 (C–O–C), 1109 (C–O), 1055 (C–O), 1031 (C–O), 891 (O–Canom–H), 656. 13C NMR (δ, ppm): 105.2–104.1 (C1), 88.5 (C4cryst), 83.3–81.9 (C4amorph), 74.6, 72.1, 71.3 (C2, 3, 5), 64.8 (C6cryst), 62.3 (C6amorph). XRD (°, 2θ (count number)[h k l]): 34.4 (3101)[040], 22.4 (14387)[002], 15.7 (6274)[101][10]; dp (Å): 41.9[002]; 45.2[040]. CI (%): 70.9XRD-PHR; 48.8XRD; 55.3NMR. SEC (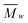 [g mol−1], PDI, DP): 112615 (8.37) 695. Elemental analysis, experimental (%): C, 41.31; H, 5.90. Calculated: for anhydroglucose (C6H10O5)%: C, 44.45; H, 6.22; and for glucose (C6H12O6)%: C, 40.00; H, 6.71.
[g mol−1], PDI, DP): 112615 (8.37) 695. Elemental analysis, experimental (%): C, 41.31; H, 5.90. Calculated: for anhydroglucose (C6H10O5)%: C, 44.45; H, 6.22; and for glucose (C6H12O6)%: C, 40.00; H, 6.71.
Results and discussion
Extraction/reaction continuous methodology
Herein we report a one-pot green and optimized extraction/reaction methodology to obtain highly pure cellulose polymers from agar industry residues. Furthermore, the extraction/reaction procedure is facilitated as the residues are based in red seaweed, which is devoid of recalcitrant lignin. The Cel source used here is considered sustainable, as it comes from the residue of algae that can be cultured. The developed method also appears to be more efficient and environmentally friendly, as the following sections clearly describe improvements in these areas. A concomitant extraction/reaction system to obtain pure cellulose from these residues first of all reduced the overall process time, also minimizing the amount of solvent used for it, which was only the volume of the solvent container in the experimental set up. The extraction/reaction equipment (normally used for liquid–liquid extraction) was here used also as a two flask apparatus. Flask 1 (a1 in Scheme 1), the extraction body, is the extraction/reaction solid–liquid flask, where the agar wastes are placed and pure cellulose polymers remain at the end. Flask 2 (c in Scheme 1), collecting round bottom flask, is the liquid–vapour flask, where the extracted/reacted contaminants are concentrated and also feeds Flask 1 with freshly distilled solvent, through its evaporation. With this configuration, a continuous extraction of Cel contaminants is ensured by displacement with freshly distilled solvent. The equilibration of the system between these two flasks is achieved through a solvent return line equipped with a Teflon® valve (a3 in Scheme 1). The continuous system equilibrates while the dissolution–evaporation steps are carefully maintained. Moreover, this solvent can be recovered without the need of further distillation or purification. The energy used is also assumed to be little, because of the time reduction of the overall process, due to the preconcentration of all contaminants. Added to this, the physicochemical properties of Cel were influenced by changes in the extraction/reaction procedure. Two steps of the treatment were modified by altering the nature of the extraction solvent using an acid or alkali. The importance of this simple strategy to obtain Cel polymers from an industrial residue is highlighted in the following sections.Characterization of the agar industries residues
The 13C CP-MAS NMR spectrum, see Fig. 1, of the washed agar industry residues displays signals corresponding to the six carbon atoms of the anhydroglucose repeating unit of Cel in chemical shifts (δ, ppm) between 110 and 60 ppm. Following the conventional numbering of the carbon atoms in the ring, the signal assignment to each carbon is given as follows: at 110–100 ppm, C1 appeared as the most frequency shifted signal due to the connectivity of this carbon with two attached oxygen atoms (as –OR groups); the signal at 90–80 ppm was assigned to C4 (tertiary carbon with one –OR); the cluster of resonances at 78–70 ppm were attributed to tC5 (tertiary carbon with one –CH2OH and one –OR), C2 and C3 (both tertiary carbons with one hydroxyl substituent), and, finally, the signal at 68–60 ppm was attributed to C6.31,40 In addition, a signal in 180–168 ppm, marked as C7, was observed and attributed to amide carbonyl nuclei due to the presence of proteic material. Some small intensity signals are also present in 135–127 ppm, marked as C8, and attributed to aromatic carbon nuclei present again in proteic material. Other signals characteristic of alpha carbon nuclei substituted with electronegative heteroatoms appeared overlapped in the polysaccharide counterpart region, primarily in the 70–45 ppm range. Those signals appearing at 45–8 ppm, marked as C9, belong to aliphatic quaternary, tertiary, secondary and primary carbon nuclei.33,41,42 As has been stated in the literature, the last described signals are related to an approximate 25% of protein content in the organism.33,41,43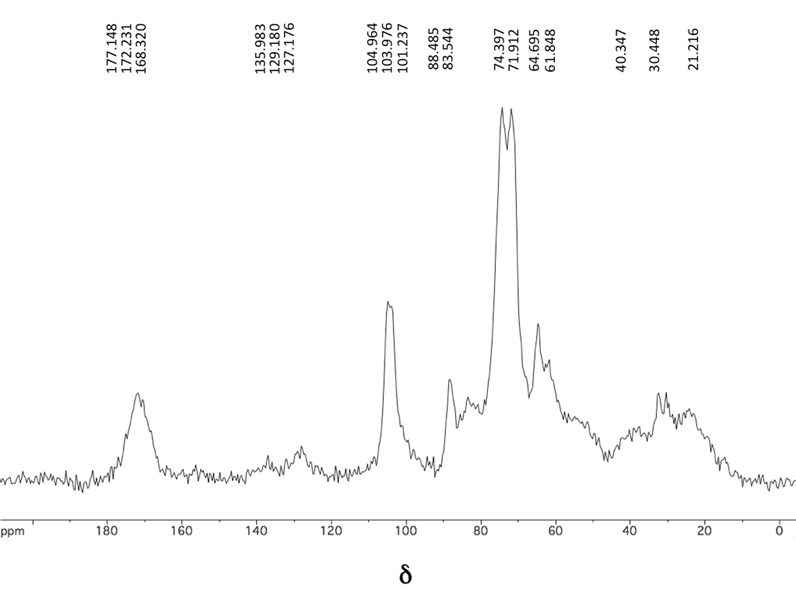 | ||
| Fig. 1 13C CP-MAS NMR spectrum of agar residues. | ||
The FT-IR spectra of the residues were obtained and used as references, thus these spectra are placed in the lowest parts of Fig. 2a, b and c. Three characteristic Cel bands were observed in the spectrum at the following frequencies (ν, cm−1) 3500, 2900 and around 1050, which are attributed to O–H, C–H and C–O respectively. The band at 898 cm−1 corresponding to the O–C–H interaction in the anomeric carbon.44,45 Additionally, the smaller bands at 1648 (COamide-I), 1430, 875 and 712 cm−1 have shown the presence of other by-products and impurities; in our case, those bands correspond to amides of proteic content,46 as well as carbonates (calcite) and silicates.47 The presence of calcite in the agar residues is attributed to the calcareous exoskeleton that protects the red algae thalli from the environment.6,48
 | ||
| Fig. 2 FT-IR spectra of agar residues and Cel samples from: neutral treatment (a), acidic treatment (b) and basic treatment (c), in each of the extraction steps (from bottom to top: residue sample, sample after methanolic wash, sample after bleaching, sample after basic wash, sample after the acidic wash, commercial standard). | ||
According to the CHNS elemental analyses, the algae residues have shown 61.66 (C), 8.51 (H), 6.09 (N) and 0.27 (S) percent contents, thus confirming the presence of sulfolipids and proteins previously identified in the NMR and FT-IR spectra.49,50 The C, H, N and S content from other lignocellulosics like leaves, sugar cane bagasse, garden residues and even paper are of the order of 44, 6, 1.5 and 0.3%, respectively.51,52 The algae residues have a higher C content, therefore a higher amount of Cel is present, compared with the content in seaweed, both in red and green algae, which is thus nearly 30%.53,54
Likewise, the XRD diffractogram of the residues was determined and used as a reference, thus it is placed in the lowest part of Fig. 3a, b and c. The diffractogram exhibited the diffraction pattern corresponding to MCC with the characteristic peaks at approximately 15, 17.3, 20.1, 23 and 35 2 θ angles (°) due to the major reflective planes in MCC as given by the Miller indices [h k l], [101], [10], [002], [021] and the diatropic planes [040]. The diffractogram displays the paratropic plane [002] typical of crystalline Cel, showing the most prominent reflections and providing evidence of the most repeated plane within the crystallite structure. Additionally, the merged diffraction peaks [101] and [10] and a [040] weak peak were also indexed.35,45 Other peaks were observed near 23° and 30° and above 35° in the residues sample and were identified as silicate and carbonate phases,47 confirming the presence of these impurities as it was already observed by FT-IR.
![Representative XRD profiles of agar residues and Cel samples from: neutral treatment (a), acidic treatment (b) and basic treatment (c), in each of the extraction steps (from bottom to top: residue sample, sample after methanolic wash, sample after bleaching, sample after basic wash, sample after the acidic wash, commercial standard). In all cases, the characteristic peaks for the extracted Cel were 15° ([101] plane), 17.3° [101̄], 20.1 ([021] plane), 23° ([002] plane) and 34.5° ([040] plane).](/image/article/2012/RA/c2ra22185c/c2ra22185c-f3.gif) | ||
| Fig. 3 Representative XRD profiles of agar residues and Cel samples from: neutral treatment (a), acidic treatment (b) and basic treatment (c), in each of the extraction steps (from bottom to top: residue sample, sample after methanolic wash, sample after bleaching, sample after basic wash, sample after the acidic wash, commercial standard). In all cases, the characteristic peaks for the extracted Cel were 15° ([101] plane), 17.3° [10], 20.1 ([021] plane), 23° ([002] plane) and 34.5° ([040] plane). | ||
Structural characterization of the Celn, Cela and Celb polymers
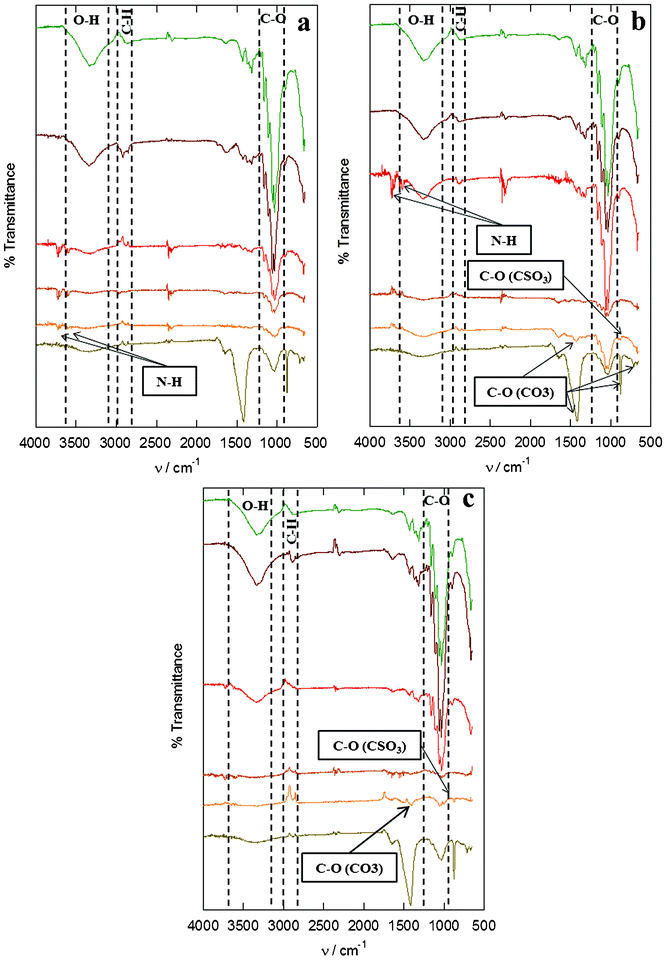
The 13C NMR spectra of the extracted Celn, Cela and Celb polymers, see Fig. 4, show the signals corresponding to the six carbons related to the monomeric unit of Cel (anhydroglucose) at chemical shifts (δ, ppm) 105–103 for C1, 90–78 for C4, 74–71 for C2, C5 and C3, and finally 64–62 for the signal corresponding to C6.31,40 In comparison to the NMR spectra of agar residues, no additional signals were observed, providing enough evidence that the employed reaction/extraction procedure led to pure Cel polymers.14,36,39 The crystalline as well as the amorphous phases of the material can be distinguished from the well defined signals at 90–85 and at 85–78, respectively. These resonances are discussed in detail in the following section on the analysis of crystallinity.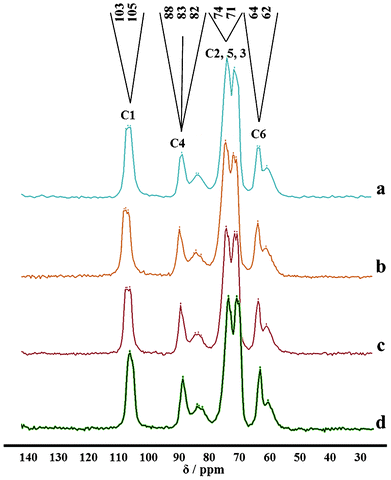 | ||
| Fig. 4 13C CP-MAS NMR spectra of Cel from: neutral treatment (a), acidic treatment (b), basic treatment (c) and commercial standard (d). The order of the six carbons was established in Fig. 1. | ||
After the final extraction step, the FT-IR spectra of the samples from the three different polymers conform to the structure of Cel, displaying the characteristic bands of functional groups present in the molecule at frequencies (ν, cm−1) of 3500, 2900 and 1050 for the O–H, C–H and C–O, respectively.45,47,55
Furthermore, the spectra, see Fig. 2, show the effect of each of the steps (methanolic wash, bleaching, basic wash and acidic wash) of the extraction process for the three polymers (Fig. 2a for Celn, 2b for Cela, and 2c for Celb). The spectra for all the polymers show that the methanolic wash eliminates the calcites and silicates (1430, 875 and 712 cm−1). It is after this step that the signals of the sulfolipids from the algae residues (C–O bond belonging to sulfate ester bands at 980 and 935 cm−1) are obvious for the samples from Celb and Cela.56 These bands are not present in the Celn sample. During the bleaching, a color change in the sample was observed, as expected, due to the removal of residual pigments still present in the agar residues.57 The bleaching with sodium chlorite removes the pigments through the formation of chlorine dioxide, a highly oxidizing agent which causes minor degradation of the Cel under the weak acidic conditions of the acetate solution.58 The basic washing step leads to the swelling of the material, increasing the volume and exposing more surface area to allow for removal of low molecular weight species.56,59 The bands associated with sulfolipids (C–O) are eliminated following this basic washing.54 After the methanolic washing, two double bands at 3600 and 3750 cm−1 are identified in the FT-IR spectra of all polymers; these N–H bands are related to proteins in the algal residue in accordance with the NMR analysis. During the final acidic washing, the residual proteins (bands with two peaks at 3600 and 3750 cm−1) in the samples were removed, obtaining pure Cel in all treatments.60,61
According to the CHNS elemental analyses, the C and H contents (%) in Celn, Cela and Celb were around 41 and 5.85, whereas for the MCC the C and H contents (%) were 42 and 6, respectively. The amount calculated for anhydroglucose (C6H10O5)% was C, 44.45 and H, 6.22, and for glucose (C6H12O6)% was C, 40.00 and H, 6.71. The absence of N and S in the chromatograms confirmed the removal of proteins and pigments in the Celn, Cela and Celb samples, and was thus in accordance with the FT IR and NMR results.62
The XRD profiles of MCC and the extracted samples are shown in Fig. 4. The diffractograms of the extracted samples show in all cases all the major reflective planes in Cel: a narrow peak for the paratropic plane 23° [002], typical of crystalline Cel, as the most prominent reflection, a diffuse peak between 13° and 18° [101] and [10], and the diatropic planes 35° [040]. The impurities (silicates and carbonates) that contributed to the signals between 23° and 30° and above 35° from the residues were eliminated through the extraction procedure during the methanolic wash.24,35 Moreover, the samples from the Cela and Celn polymers show a change in crystallinity, through the action of NaOH during the basic wash, as has been previously reported for other Cel samples.63 However, mercerization during this basic washing is not likely, due to the fact that the diffractograms do not show the characteristic peaks (12.5°, 20° and 22°) for Cel II.64,65 After the acidic wash, the peaks in the diffractograms of the samples from the three polymers corresponded to those of MCC.66
Yield, CI, dp, MW and PDI of the extracted Cel polymers
Table 1 below shows the results from the characterization of the extracted samples. The highest yields, approximately 433 mg Cel per g residue (72.2% glucose based) and 420 mg Cel per g of residue (70% glucose based) were obtained from the basic and neutral treatments (Celb and Celn polymers) respectively. The lowest Cel yield, approximately 30 mg Cel per g of residue (56.7% glucose based), was obtained from the acidic treatment (Cela). These yields can be compared with other residues provided from superior plants, like oil palm trunk (45.3%)67 or sugar cane bagasse (45.9%).68 The yield obtained is higher compared to other red seaweed, such as Gelidiella acerosa (13.65% ± 0.5%), Gelidium pusillum (9.3% ± 0.4%), Gracilaria edulis (5.3% ± 0.2%).29| Sample | Gravimetric yield [mg Cel per g residue] | Yield%a | CIXRD-PHR | CIXRD | dp [Å] | CINMR |
|
PDIb | DP | |
|---|---|---|---|---|---|---|---|---|---|---|
| (0 0 2) | (0 4 0) | |||||||||
a Yields were calculated on the basis of amount glucose (58.1%) present in red algae Gelidium after agar extraction obtained by Vigon, et al. in 1994.39
b
 is the number-average molar mass. is the number-average molar mass.
|
||||||||||
| MCC | — | — | 77.7 | 56.6 | 50.3 | 48.8 | 58.6 | 61673 |
5.33 | 381 |
| Cela | 340 | 58.6 | 72.6 | 51.2 | 45.1 | 53.1 | 59.3 | 78856.5 |
7.22 | 487 |
| Celn | 420 | 72.4 | 72.8 | 47.4 | 40.1 | 38.0 | 55.7 | 148527.5 |
11.82 | 917 |
| Celb | 433 | 74.6 | 70.9 | 48.8 | 41.9 | 45.2 | 55.3 | 112615 |
8.37 | 695 |

The CI from the XRD peak height ratio method (CIXRD-PHR) of all samples, including the standard, was approximately 15% higher than that calculated for other methods. This method is the most widely used for the calculation of Cel CI.35,69 The CIXRD-PHR (> 70%) of the Cel obtained from agar residues was higher compared to reported values for Cel polymers from other red algaes like Gelidae acerosa (66%), Gracilaria textoria (64%) or Gracilaria debilis (66%).28 However, it was lower compared to Cladophora sp. (95.7%).24 The overestimation in the calculation of the CIXRD-PHR compared with the CIXRD is attributed to the fact that the first method considers only one peak [002], compared with the second method (CIXRD) where crystalline and amorphous regions are considered for the estimation (Table 1).35 With this last method, Cela had the highest crystallinity index (51.2%). Crystallite size, both in thickness [002] and in length [040], was 45.1 and 53.1 Å respectively. These crystallites can be considered highly crystalline nanostructures,70 with cuboid form. Celn and Celb have almost the same CIXRD, 47.4 and 48.8%. The dp of Celn indicates a cubic form where the thickness and length are 40.1 and 38 Å. Finally, Celb is slightly longer with 45.2 Å and 41.9 Å thickness. The thickness of all extracted polymers was in the order of 43 ± 2 Å. In contrast, wood powder is about 32 ± 0.6 Å, Chaetomorpha melagonium 35 Å and cotton 40 ± 10 Å. However, the length of the crystallites in these species is 280, 170 and 70 Å.71–73 Therefore, the Cel extracted forms from agar residues all have a particularly cuboid form.
The CI of the extracted Cel polymers calculated by NMR (CINMR) (see Table 1) were in all cases higher than the ones calculated by XRD (CIXRD). This difference suggests that the NMR determination is the most accurate in calculating crystallinity, since it provides well-separated or distinguishable signals to quantify both amorphous or crystalline phases present. The CINMR of the samples ranged from 55 to 59%, compared to around 47 to 51% for the CIXRD. As with the previous method, Celn and Celb had equivalent crystallinity. However, the samples from the Cela showed the highest crystallinity of all extracted samples and a slightly higher crystallinity than the commercial sample. The high crystallinity of Cela may be attributed to the action of HCl with water at low temperatures, causing a partial recrystallization of the biopolymer. In addition, the solution protonates the glycosidic linkages closing the loose links in an ordered manner, forming hydrocellulose, which nonetheless has a higher crystallinity.66,74
Celn had the highest molecular weight, almost double that of Cela and the commercial sample. This is attributed to the milder (neutral) conditions during the extraction. Alkaline degradation through the breakage of glycosidic bonds occurs at temperatures around 140 °C. However, in the conditions used for the Celb, the alkaline hydrolysis of Cel is not probable, and the reduction in the chain length compared to the Celn is attributed to peeling or erosion of monomer units.66Cela had the lowest molecular weight and polydispersity. This could be due to the fact that the HCl, in combination with methanol during the methanolic wash, is introduced into the glycosidic linkage from the amorphous structure of the polymer matrix, causing chain rupture and a consequently lower molecular weight.75,76 All samples were broad, having polydispersities from 7.22 to 11.28, which are higher than that for MCC.
The DP of extracted Cel were 917, 487 and 695 for Celn, Cela and Celb, respectively, which are all higher than the DP values for MCC (381), cotton purified (250), Hemp (350) and various commercial and fibrous Cel (270).77,78
With the minimal solvent extraction/reaction system it is possible to obtain pure Cel from agar residues in a short time and using small amounts of all solvents,27,28, thus we have an efficient, environmentally friendly method for Cel extraction.
Conclusions
Pure Cel polymers (devoid of lignin) were obtained from algae residues in a continuous extraction/reaction experimental device, under neutral (Celn), acidic (Cela) or basic (Celb) treatments, followed by neutralization, all performed in situ with minimum solvent use and during diminished extraction/reaction periods. Yields (%) were as high as 43.3 (mg extracted Cel per g residue) or 72.2 (based on a 60% carbohydrate content present in residues), and resulted higher in comparison to other described methodologies.The Celn, Cela and Celb polymers were characterized by 13C CP-MAS, NMR, FT-IR, CHNS elemental analyses, XRD and SEC techniques, all giving experimental insights into the nature of the isolated materials. The NMR and FT-IR spectra of the Celn, Cela and Celb polymers confirmed their chemical integrity, while the elemental analyses have shown a successful removal of pigments and proteins.
X-ray diffractograms corresponded to the pattern obtained for MCC. The CI (%) (XRD/CP-MAS NMR) of the extracted materials was determined as 47.4/55.7 for Celn, 51.2/59.3 for Cela and 48.8/53.3 for Celb. The CI for all the samples calculated by NMR resulted in slightly higher values in comparison to XRD. In general, these procedures provided Cel polymers of medium crystallinity. Cela (45.1 and 53.1 Å) can be considered to be formed of highly crystalline nanostructures, with cubioid form, while Celn has a cubic form (40.1 and 38 Å) and Celb is slightly longer (45.2 Å and 41.9 Å).
The 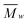 [g mol−1] and PDI were 148528 (11.82) for Celn, 78857 (7.22) for Cela, 112615 (8.37) for Celb, and 61673 (5.33) for MCC. The corresponding DP was 917 for Celn, 487 for Cela, 695 for Celb and 381 for MCC. The mildest procedure was the neutral treatment, since it provides Celn with the highest molecular weight. Nevertheless, the PDI in Celn is the highest in the series.
[g mol−1] and PDI were 148528 (11.82) for Celn, 78857 (7.22) for Cela, 112615 (8.37) for Celb, and 61673 (5.33) for MCC. The corresponding DP was 917 for Celn, 487 for Cela, 695 for Celb and 381 for MCC. The mildest procedure was the neutral treatment, since it provides Celn with the highest molecular weight. Nevertheless, the PDI in Celn is the highest in the series.
The acidic treatment provided Cela, which was the most crystalline material with the smallest and narrowest molecular weight distribution (53% of Celn). The basic treatment provided Celb, which was the highest in yield in the series, with a high degree of polymerization (75.8% of Celn) and maintaining a crystallinity very similar to that of Celn. The characteristics of Celn should be closely related to those present in untreated Cel, as it is synthesized and used in the natural source.
This minimal solvent extraction/reaction system can be used for Cel extraction from other residual sources, in order to obtain an added value final product while caring for the environment, as it is a method that uses both low quantities of solvents and energy costs, due to its continuous nature. On the other hand, depending on the final application for Cel materials, the isolation procedures provided herein could serve as starting points for types of Cel polymers with different characteristics, and also differing from those of MCC, to be studied or employed in potential applications.
Acknowledgements
We thank Agarmex and Oceanographer David Lora for the kind donation of the agar residues samples. Roxana López Simeon thanks CONACyT for a postgraduate scholarship no. 34852. José Campos-Terán is grateful with CONACyT (CB-2007/83535), PROMEP (UAM-PTC-060) and UAM-Cuajimalpa (Acuerdo 01/2007) for funding provided to develop part of this work. Hiram I. Beltran is grateful with CONACyT (CB82255/105532) and TWAS (07-228 RG/CHE/LA) for funding provided to develop part of this work. Maribel Hernández-Guerrero thanks CONACyT (CB 134267/47410235) and PROMEP (UAM-PTC-140) for funding to conduct this research. We thank Dr Ernesto Rivera Becerril (DCN, DCNI, UAM, Unidad Cuajimalpa) for kind discussions to understand some points of the current research. All the authors of this contribution acknowledge support provided from “Retos y oportunidades del aprovechamiento de la biomasa lignocelulósica para la obtención de biocombustibles y otros productos de valor agregado en México” Project Funded from UAM-C (Acuerdo 01/2011). The authors are also grateful to Dr Alberto López Luna, Dr Keiko Shirai Matsumoto (UAM, Unidad Iztapalapa) for their kind help with the GPC and Manuel Aguilar Franco (UNAM) for XRD collection.References
- P. A. Faugeras, P. H. Elchinger, F. Brouillette, D. Montplaisir and Z. Rachida, Green Chem, 2012, 14, 598–600 RSC.
- B. Zhao, L. Greiner and W. Leitner, RSC Advances, 2012, 2, 2416–2479 Search PubMed.
- F. J. Ruiz-Dueñas and A. T. Martínez, Microbiology Biotechnology, 2009, 2, 164–177 CrossRef.
- R. E. Texeira, Green Chem., 2010, 14, 419–427 RSC.
- J. González, Ciencias., 1987, 10, 16–25 Search PubMed.
- C. J. Dawes, Botánica Marina, 1991. Ed. Limusa. México Search PubMed.
- O. A. Vian, and G. A. Brusi, Introducción a la química industrial, 1999, Ed. Reverté, Barcelona Search PubMed.
- N. A. Campbell, J. B. Reece, Biología, 2006, 7th edn,Pearson Education, CA. Search PubMed.
- R. A. Melo, B. W. W. Harger and M. Neushul, Hydrobiologia, 1991, 221, 91–106 CrossRef.
- A. Tye, M. Fullen and T. Hocking, Community Soil Scientific Plant Analysis, 2001, 32(3&4), 311–329 CAS.
- K. Okuda, K. Oka, A. Onda, K. Kajiyoshi, M. Hiraoka and K. Yanagisawa, Journal of Chemical Technology and Biotechnology, 2008, 83, 836–841 CrossRef CAS.
- B. Santelices, Hydrobiologia, 1991, 221, 31–44 CrossRef.
- S. A. Guzmán del Próo, Ciencia Pesquera, 1993, 9, 129–136 Search PubMed.
- M. C. Rodríguez, M. C. Matulewicz, M. D. Noseda, D. R. B. Ducatti and P. I. Leonardi, Bioresource Technology, 2009, 100, 1435–1441 CrossRef.
- J. B. Harris and H. Porse, Journal of Applied Phycology, 2010 DOI:10.1007/s10811-010-9529-3.
- Comisión Nacional de Acuacultura y Pesca (CONAPESCA), Anuario Estadístico, 2010.
- Sistema Nacional de Información Arancelaria (SNCI-Economía) 2011http://www.economia-snci.gob.mx:8080/siaviWeb/siaviMain.jsp.
- Y. Freile-Pelegrín, D. Robledo and E. Serviere-Zaragoza, Hydrobiologia, 1999, 398(399), 501–507 CrossRef.
- E. Serviere-Zaragoza, D. Rodríguez-Vargas and J. González-González, Hydrobiologia, 1993, 260(261), 45–50 CrossRef.
- R. M. Cubillos, Revista de Biología Marina, 1950, 3(1-2), 70–88 Search PubMed.
- C. C. Stanford, American Journal of Pharmacy, 1983, 55(12), 1–16 Search PubMed.
- H. Mertaniemi, A. Laukkanen, J. E. Teirfolk, O. Ikkala and R. H. Ras, RSC Advances, 2012, 2, 2882–2886 RSC.
- D. Rajalaxmi and A. J. Ragauskas, RSC Advances., 2012, 2, 3403–3409 RSC.
- A. Mihranyan, A. Piñas Llagostera, R. Karmhag, M. Strømme and R. Ek, International Journal of Pharmaceutics., 2004, 269, 433–442 CrossRef CAS.
- R. Ek, C. Gustafsson, A. Nut, T. Iversen and C. Nyström, Journal of Molecular Recognition., 1998, 11, 263–265 CrossRef CAS.
- J. Curvers, T. Noij, C. Cramers and T. Rijks, Chromatographia, 1984, 19, 225–230 CAS.
- B. Stachel, K. Baetjer, M. Cetlnkaya, J. Duesreln, U. Lahl, K. Llerse and W. Thiemann, Analytical Chemistry, 1981, 53(9), 1469–1472 CrossRef CAS.
- J. Slayton, S. Warner, P. Shreiner, C. Tulip and E. Messer, EPA/903/R-93/003, 1993 Search PubMed.
- Environmental Monitoring Systems Lab O. H. Cincinnati, EPA/46724505, 1997 Search PubMed.
- W. D. P. Stewart, Algal Physiology and Biochemistry. Blackwell Scientific. Oxford. 1974 Search PubMed.
- A. K. Siddantha, P. Kamalesh, M. Ramavatar, P. Gayatri, K. M. Gaurav, U. C. Mahesh, D. O. Mihir, K. Sanjay and D. Naresh Sanandiya, Bioresource Technology, 2009, 100, 6669–6673 CrossRef.
- A. K. Siddhanta, U. Mahesh Chhatbar, K. Gaurav Mehta and D. Naresh Sanandiya, J Appl Phycol, 2011, 23(5), 919–923 CrossRef CAS Sanjay Kumar & Mihir D. Oza & Kamalesh Prasad & Ramavatar Meena..
- M. R. Vignon, C. Rochas, R. Vuong, P. Tekely and H. Chanzy, Botanica Marina, 1994, 37, 331–340 Search PubMed.
- J. Schaefer and E. O. Stejskal, Journal of American Chemistry Society, 1976, 98(4), 1031–1032 CrossRef CAS.
- S. Park, J. O. Baken, M.E. Himmel, P. A. Parilla and D. K. Johnson, Biotechnology for Biofuels, 2010, 3, 1–10 CrossRef.
- R. H. Newman, Holzforschung, 2004, 58, 91–96 CrossRef CAS.
- J. Xiao-Juan and D. Pascal Kamdem, Cellulose Chem. Technol., 2009, 43(7-8), 229–234 Search PubMed.
- A. F. Turbak, A. El-Kafrawy, F. W. Snyder, A. B. Auerbach, 1981, Solvent systems for cellulose, U.S. Patent 4 302,252 Search PubMed.
- E. Sjöholm, Size Exclusion Chromatography of Cellulose and Cellulose Derivatives, In: Wu, C.S. (ed.), Handbook of Size Exclusion Chromatography and Related Techniques, Chromatographic Science Ser., 2004, Marcel Dekker, New York Search PubMed.
- R. L. Dudley, C. A. Fyfe, P. J. Stephenson, Y. Deslandes, G. K. Hamer and R. H. Marchessault, Journal of American Chemistry Society, 1983, 105(8), 2469–2472 CrossRef CAS.
- M. A. Hurtado, M. Manzano-Sarabia, E. Hernández-Garibay, I. Pacheco-Ruiz and J. A. Zertuche-González, Journal of Applied Phycology, 2011, 23, 727–734 CrossRef.
- M. R. Vignon, E. Morgan and C. Rochas, Botanica Marina, 1994, 37, 325–329 CAS.
- M. Sekkal, J. P. Huvenne, P. Legrand, B. Sombret, J. C. Mollet, A. Mouradi-Givernaud and M. C. Verdus, Mikroquimica Acta, 1993, 112, 1–10 CrossRef CAS.
- X. F. Sun, R. C. Sun, P. Fowles and M. S. Baird, Carbohydrate Polymers, 2004, 55, 379–391 CrossRef CAS.
- L. Ruigang, Y. Hui and Y. Huang, Cellulose, 2005, 12, 25–34 CrossRef.
- J. Kong and S. Yu, Acta Biochimica et Biophysica Sinica, 2007, 39(8), 549–559 CrossRef CAS.
- D. Fragoso, F. Ramírez-Cahero, A. Rodríguez-Galván, R. Hernández-Reyes, A. Heredia, D. Rodríguez, M. Aguilar-Franco, L. Bucio and V. A. Basiuk, Ciencias Marinas, 2010, 36(1), 41–58 CAS.
- M. A. Borowitzka, Marine Biology, 1981, 62, 17–23 CrossRef CAS.
- L. Talarico, Scientia Marina, 1996, 60(1), 205–222 CAS.
- E. McCandless, Ann. Rev. Plant Physiol., 1979, 30, 41–53 CAS.
- G. Tchobanoglous, H. Theisen and S. A. Vigil, Gestión Integral de Residuos Sólidos, 1998, Mc Graw Hill. Search PubMed.
- P. Sugumaran and S. Seshardi, Journal of Scientific & Industrial Research, 2009, 68, 719–723 CAS.
- M. Vaher, K. Truus, I. Taurus, I. Leito, L. Lahe Kallavus and T. Pehk, Proc. Estonian Acad. Sci. Chem., 1998, 47(2), 51–59 CAS.
- M. Shanmugam, B. K. Ramavat, K. H. Mody, R. M. Oza and A Tewan, Indian Journal of Marine Sciences, 2001, 30(4), 222–227 Search PubMed.
- D. Ciolacu, F. Ciolacu and V. I. Popa, Cellulose Chemistry and Technology, 2011, 45(1-2), 13–21 CAS.
- G. L. Mayers, M. Pousada and T. H. Haines, Biochemistry, 1969, 8, 2981–2986 CrossRef CAS.
- M. C. Taylor, J. F. White, G. P. Vincent and G. L. Cunningham, Industrial and Engineering Chemistry, 1940, 32(7), 899–903 CrossRef CAS.
- A. R. George and E. G. Kenneth, Industrial & Engineering Chemistry, 1940, 32(8), 1122–1128 Search PubMed.
- D. Horton, Advances in Carbohydrate Chemistry, 1964, 19, 256–260 CrossRef.
- J. N. C. Whyte and J. R. Englar, Phytochemistry, 1981, 20(2), 237–240 CrossRef CAS.
- D. R. Viera-Oramas, Z. Zhurbenko and C. Rodríguez-Martínez, Revista Cubana de Medicina Tropical, 2009, 61(2), 1–10 Search PubMed.
- M. Seifert, S. Hesse, V. Kabrelian and D. Klemm, Journal of Polymer Science Part A: Polymer Chemistry, 2003, 42(3), 463–470 CrossRef.
- M. I. Felisberti and R. Stadler, Macromolecules, 1989, 22, 4130–4132 CrossRef.
- A. Isogai and R. H. Atalla, Cellulose, 1994, 5, 309–319 CrossRef.
- S. Borysiak and B. Doczekalska, Fibres & Textiles in Eastern Europe, 2005, 13(5), 87–89 CAS.
- J. L. Wertz, O Bédué and J. P. MercierCellulose Science and Technology, 2010, EPFL press, Italy Search PubMed.
- S. RunCang, J. M. Fang, L. Mott and J. Bolton, Journal of Wood Chemistry and Technology, 1999, 19(1-2), 167–185 CrossRef.
- J. X. Sun, X. F. Sun, H. Zhao and R. C. Sun, Polymer Degradation and Stability, 2004, 84, 331–339 CrossRef CAS.
- J. I. Morán, V. A. Alvarez, P. V. Cyras and A. Vázquez, Cellulose, 2008, 15, 149–159 CrossRef.
- M. Ciprian, A. Cirtiu, F. Dunlop-Briére and A. Moores, Green Chem, 2011, 13, 288–291 RSC.
- S. Andersson, R. Serimaa, T. Paakkari, P. Saranpää and E. Pesonen, J Wood Sci, 2003, 49, 531–537 Search PubMed.
- A. D. Wilson, B. E. Kent, R. J. Mesley, R. P. Miller, D. Clinton and K. Fletcher, Nature, 1970, 225, 273–274 CrossRef.
- A. V. Moharir and P. Kiekens, Journal of Applied Polymer Science, 1998, 68(13), 2107–2112 CrossRef CAS.
- L. Yan and Z. Gao, Cellulose, 2008, 15, 789–796 CrossRef CAS.
- S. Leon and M. Nelson, Textile Research Journal, 1954, 76, 4626–4630 Search PubMed.
- J. H. Lin, Y. H. Chang and Y. H. Hsu, Food Hydrocolloids, 2009, 23, 1548–1553 CrossRef CAS.
- A. Battista, S. Coppicic, J. A. Howsmon, F. F. Morehead and W.A. Sisson, Industrial and Engineering Chemistry, 1956, 48(2), 333–335 CrossRef.
- Y. H. Percival Zhang and L. R. Lynd, Biomacromolecules, 2005, 6, 1510–1515 CrossRef.
| This journal is © The Royal Society of Chemistry 2012 |