DOI:
10.1039/C2RA21288A
(Paper)
RSC Adv., 2012,
2, 11850-11857
Structural evolution in a biphasic system: poly(N-isopropylacrylamide) transfer from water to hydrophobic ionic liquid†
Received
26th June 2012
, Accepted 30th September 2012
First published on 29th October 2012
Abstract
The structural evolution of payload transfer in biphasic systems is significant but difficult to accurately follow over the whole process. The dynamic transfer of homopolymer poly(N-isopropylacrylamide) (PNIPAM) from water to a hydrophobic IL (1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide, [C2MIM][NTf2]) phase was investigated by in situ react infrared (IR) spectroscopy to reveal the detailed transfer process and structural changes of PNIPAM, water and [C2MIM][NTf2]. Compared with the corresponding binary systems, this work summarizes the four-step transfer behavior and the interactions among the components according to IR spectroscopy and multivariate curve resolution (MCR). At the initial stage, only some water diffuses into the hydrophobic IL in the form of a symmetric 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2-type hydrogen bonded (anion⋯H2O⋯anion) species while most of the PNIPAM molecules are preserved in the water layer in the form of aggregates. Afterwards, the water molecules are bonded with PNIPAM in the aggregated state and they start to transfer together into the IL layer in the second stage where PNIPAM is changed into unfolding structures with complex interactions such as H2O⋯PNIPAM⋯anion, and meanwhile water structures of anion⋯(H2O)n⋯anion form due to further diffusion of water. After these stages, water molecules continue to dissolve in the IL although the polymer transfer is completed, and they are inclined to form further water clusters (H2O)n as deduced from the increase of OH region (3500–3300 cm−1) absorption. Finally, all of the components (PNIPAM, water and IL) coexist in the homogeneous IL phase with the aid of complex hydrogen bond and electric charge interactions.
:2-type hydrogen bonded (anion⋯H2O⋯anion) species while most of the PNIPAM molecules are preserved in the water layer in the form of aggregates. Afterwards, the water molecules are bonded with PNIPAM in the aggregated state and they start to transfer together into the IL layer in the second stage where PNIPAM is changed into unfolding structures with complex interactions such as H2O⋯PNIPAM⋯anion, and meanwhile water structures of anion⋯(H2O)n⋯anion form due to further diffusion of water. After these stages, water molecules continue to dissolve in the IL although the polymer transfer is completed, and they are inclined to form further water clusters (H2O)n as deduced from the increase of OH region (3500–3300 cm−1) absorption. Finally, all of the components (PNIPAM, water and IL) coexist in the homogeneous IL phase with the aid of complex hydrogen bond and electric charge interactions.
Introduction
Studies of the structural evolution of molecules in solution are essential for understanding the relationship between the structure and properties of polymers. For example, a flexible polymer chain can resemble a molten globule or an aggregate as the solvent becomes poor. Those polymer associations, however, can undergo a “globule-to-coil” transition or dissolve in the solution again when its external environment changes.1–5 The dissolution abilities also usually change with the molecule structures. In addition, the transition between the unfolding and folding structures is also an important aspect in biochemical applications such as protein denaturation6–8 and drug release.9–11
Poly(N-isopropylacrylamide) (PNIPAM) is a widely used biochemical material due to its rapid thermal response,12–15 and it is also used as a model to study different chain conformations. PNIPAM exhibits a separation transition above its lower critical solution temperature (LCST) at 32 °C in aqueous solution as a result of hydrophobic interaction and amide–amide hydrogen bonds, which has been extensively studied by means of turbidimetry,16–19 differential scanning calorimetry,20–22 fluorescence,23–25 light scattering,26–28 nuclear magnetic resonance29–31 and Fourier-transform infrared spectroscopy (FTIR).32–35 Furthermore, the dissolution behavior of PNIPAM chains was also observed in a hydrophobic ionic liquid (IL), 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([C2MIM][NTf2]) above the upper critical solution temperature (UCST).36 Moreover, in our previous researches we found that its phase transition was mainly caused by the interaction between NH groups of PNIPAM and the anion of [C2MIM][NTf2].37
Due to the opposite dissolution abilities of PNIPAM in these two solvents, polymer chains can transfer from water to IL with unfolding structure at an elevated temperature. Therefore, such a biphasic system can be used to develop a convenient payload delivery and release through conformational changes of polymer chains.38–41 However, clarification of how the polymer is transferred in the biphasic system and the structures adopted during the diffusion is required. In the literature, light scattering,38–40,42,431H NMR spectroscopy,40,43 and UV-vis spectroscopy40,41,44 have been used to characterize the micelle size, transport efficiency after payload transfer, but the dynamic changes of the complex structures involving ternary composites (polymer, water and IL) in the whole process are not clear. Moreover, Kazarian et al. found most ILs based on imidazolium cations with different anions show hydrogen bonding with water molecules,45 so it is inevitable that the interaction among ternary composites such as polymer–water, polymer–IL and water–IL will influence the structures of polymersomes.
It is well known that IR spectroscopy is very suitable for monitoring the changes of specific groups assigned to different components, and several molecular interactions can easily be monitored from the variation of intensities and wavenumbers.46,47 Furthermore, ATR-FTIR has previously been successfully applied to analyze many diffusion systems.48–51 Examples are extraction of impurities from ILs using supercritical carbon dioxide (scCO2)52,53 and lipase from an ionic liquid-based aqueous biphasic system.54 This methodology may solve the need for continuous and in situ collection to monitor the content and structural changes of the above three-component system during transfer.
In this work, the dynamic transfer of PNIPAM from a water layer to a hydrophobic IL ([C2MIM][NTf2]) layer was investigated by a commercial ATR-FTIR system with a silicon probe. The transfer process involving PNIPAM, water and IL can easily be monitored through infrared spectroscopy and multivariate curve resolution (MCR). Due to the effect of water diffusion, furthermore, the structural evolution of ternary composites in the IL-rich phase will be discussed in detail through second derivative spectra at different transfer times by comparison with the corresponding binary systems.
Experimental section
Materials
1-Ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([C2MIM][NTf2]) was purchased from Sigma-Aldrich Corporation (>98%). N-Isopropylacylamide (NIPAM) monomer were purchased from Tokyo Kasei Kogyo Corporation (Tokyo, Japan) and used after recrystallization. Poly(N-isopropylacrylamide) (PNIPAM) was synthesized by free-radical polymerization in tetrahydrofuran (THF) with azobis(isobutyronitrile) (AIBN) initiator, and the reaction was carried out at 60 °C for 16 h under a nitrogen atmosphere. The molecular weight of PNIPAM, Mn = 1.1 × 104, Mw/Mn = 1.6.
Infrared spectra were recorded on a ReactIR 45 m spectrometer (Mettler–Toledo) equipped with a silicon probe with a resolution of 4 cm−1 and 64 scans during the shuttle process. Before adding 1 ml 10% PNIPAM aqueous solution carefully through a syringe, the probe was immersed in a glass tube with 1 ml neat IL and fixed at a certain height to make sure the silicon probe was completely contacted with the IL phase, and then the biphasic system was placed in 80 °C oil-bath. A schematic illustration of the experimental set-up is shown in Scheme 1. Each spectrum was collected at an interval of 6 min under relatively tight sealed condition to avoid evaporation of water. The control experiment of pure water diffusing into the IL was processed by similar methods and conditions.
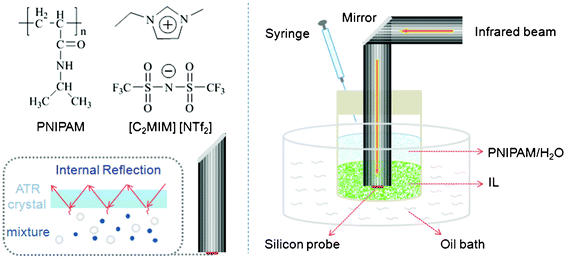 |
| | Scheme 1 Schematic illustration of the in situ ReactIR and other experimental devices. | |
Multivariate curve resolution (MCR)
MCR is an effective approach for automatically calculating pure component concentrations and spectra in complex systems.55 Herein, all spectra (from 1781 to 1590 cm−1) during the shuttle process were first obtained by subtracting the IL spectrum and baseline correcting. After the pretreatment of principal component analysis (PCA) calculation to improve the signal-to-noise ratio, multivariate curve resolution was performed to calculate the relative contents and trends by the software the Unscrambler ver. 9.7.
Results and discussion
Four-stage transfer process
Fig. 1 shows the experimental images of PNIPAM transfer from water to the IL layer. Initially, the two phases at the room temperature are transparent. PNIPAM molecules start to associate in the water phase at 80 °C while the IL layer is still clear. After homeothermic heating for a certain time, finally, the biphasic system becomes almost transparent again. However, this may not reflect the entire dynamic processes due to complicated and invisible molecule movements. As a result, the dynamic transfer was monitored by time-resolved IR spectra as water and PNIPAM molecules diffuse from the upper water layer into the IL layer at 80 °C where the changes of many bands involving PNIPAM, H2O and IL can be observed during the transfer process (Fig. 2a). Some bands of PNIPAM and H2O become difficult to identify owing to overlapped peaks of three components, so difference spectra in this region have been calculated by subtraction of pure IL infrared bands (Fig. 2b), where NH bending vibration (δNH) and OH stretching vibration (νOH) emerge clearly in the regions 1590–1490 and 3745–3000 cm−1. Additionally, it is found that the intensities of SO2 and CF3 bands in the anion of the IL are decreased slightly during transfer, which may be caused by a dilution effect as PNIPAM and water molecules transfer into the IL phase (Fig. S1, ESI†).
![Experimental images of PNIPAM transfer from H2O (upper layer) to [C2MIM][NTf2] (lower layer). The first photo is obtained at room temperature. The second photo was taken when the sample was placed in an 80 °C oil-bath for 1 min, while the third photo was taken after an isothermal process for 6 h at 80 °C.](/image/article/2012/RA/c2ra21288a/c2ra21288a-f1.gif) |
| | Fig. 1 Experimental images of PNIPAM transfer from H2O (upper layer) to [C2MIM][NTf2] (lower layer). The first photo is obtained at room temperature. The second photo was taken when the sample was placed in an 80 °C oil-bath for 1 min, while the third photo was taken after an isothermal process for 6 h at 80 °C. | |
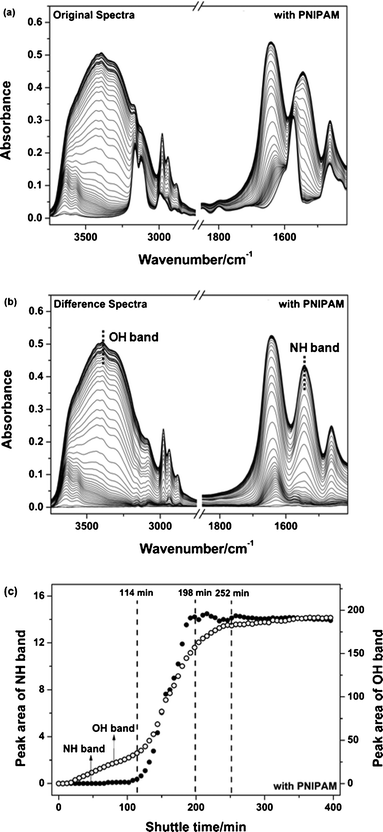 |
| | Fig. 2 Original (a) and difference (b) spectra in the region (3745–1411 cm−1) at intervals of 6 min during PNIPAM transfer process under homeothermic (80 °C) and quiescent condition. Difference spectra were obtained by subtraction of the IL IR spectrum. (c) The peak area changes of NH and OH bands as a function of transfer time based on difference spectra (●, NH band; ○, OH band). | |
To make clear the trends during the transfer, the changes of two groups (OH and NH) belonging to water and PNIPAM, respectively, were plotted as a function of transfer time in Fig. 2c. It is obvious that the NH band of PNIPAM is hardly observed in the first 114 min, which infers almost no PNIPAM transfers into the IL phase in this period. A subsequent rapid growth period (from 114 to 198 min) indicates that a significant amount of PNIPAM molecules rapidly dissolve in the IL phase. Finally, the content of PNIPAM molecules reaches a maximum and levels off after 198 min. For water, some different changes of the νOH region illustrate that a portion of water molecules gradually transfer into IL phase based on a slight increase of OH area in first 114 min. Then more water molecules diffuse during 114–252 min, which shows inconsistency with diffusion time of the PNIPAM molecules, i.e. water molecules continue to transfer into the IL phase after the transfer of PNIPAM is complete (198 min). Finally, the diffusion of water tends to stabilize by 252 min although a gentle increase is still observed. The contributions of water on transfer in the biphasic system were not noticed directly in previous shuttle systems. Recently, Lodge et al. found that the there was higher temperature of demicellization in water/[BMIM][PF6] system than in dry [BMIM][PF6] due to the fact that water is a poor solvent for PNIPAM at high temperature.38 Therefore, it is believed that the effect of water on the unfolding structure of PNIPAM in IL-rich phase should be taken into account during the transfer process.
Combined with the above photos of PNIPAM transfer in Fig. 1, therefore, the whole transfer process can be separated into four stages according to the three time points mentioned above (114, 198, 252 min). The first step before 114 min is the preparatory period where some water molecules begin to diffuse into the IL phase, while PNIPAM essentially remains in the water phase, although polymer chains are aggregated owing to phase separation in aqueous solution above the LCST. Furthermore, the changing trends of νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and δOH in the region of 1781–1590 cm−1 also support previous results that PNIPAM and water have distinct diffusion behavior before 114 min. Generally, the results of MCR show a the gentle increase of the δOH band in the IL the initial stage while the band of νCO is not changed in the first stage in Fig. 3, although their transition points are not as accurate as previous spectroscopic data, as a result of highly overlapped spectra. The second step from 114 to 198 min is the fast transfer of PNIPAM aggregations and water molecules. In this period, PNIPAM aggregations undergo globule–coil transition in the IL due to a favourable interaction (3386 cm−1) formed between NH groups of PNIPAM and the anion of the IL above the UCST (Fig. 4). The third step during 198–252 min is the rapid transfer of water alone; after PNIPAM transfer is complete, water molecules continue to diffuse into the IL phase. The fourth step after 252 min is the equilibrium of the three components, and all of them are inclined to be stabilized in both phases.
O and δOH in the region of 1781–1590 cm−1 also support previous results that PNIPAM and water have distinct diffusion behavior before 114 min. Generally, the results of MCR show a the gentle increase of the δOH band in the IL the initial stage while the band of νCO is not changed in the first stage in Fig. 3, although their transition points are not as accurate as previous spectroscopic data, as a result of highly overlapped spectra. The second step from 114 to 198 min is the fast transfer of PNIPAM aggregations and water molecules. In this period, PNIPAM aggregations undergo globule–coil transition in the IL due to a favourable interaction (3386 cm−1) formed between NH groups of PNIPAM and the anion of the IL above the UCST (Fig. 4). The third step during 198–252 min is the rapid transfer of water alone; after PNIPAM transfer is complete, water molecules continue to diffuse into the IL phase. The fourth step after 252 min is the equilibrium of the three components, and all of them are inclined to be stabilized in both phases.
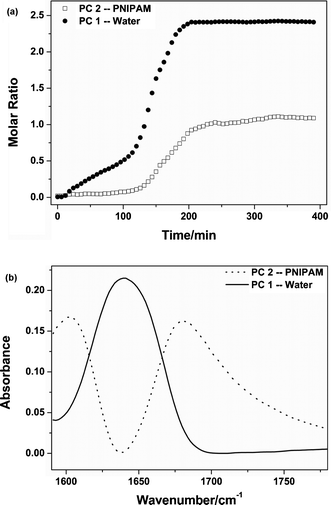 |
| | Fig. 3 Concentration profiles (a) and split spectra (b) of water and PNIPAM calculated by MCR. | |
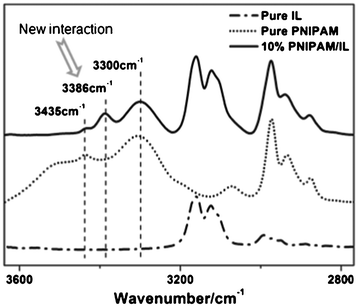 |
| | Fig. 4 Comparative FTIR spectra of neat IL, neat PNIPAM and 10 wt% PNIPAM/IL. | |
The contribution from water diffusion
It is worthwhile to note that all of transfer steps are closely related to water diffusion which is hardly noticed by turbidity measurements. Thus, it is necessary to understand the contribution from water diffusion during transfer through control experiments in the absence of PNIPAM. Fig. 5 shows the spectra and area changes of neat water diffusion under the same conditions. Three-step diffusion was observed in the control experiment. In the first 198 min, several water molecules slowly dissolve into the IL phase. The second step is rapid diffusion of water from 198 to 294 min. After that, water molecules show little further diffusion. Compared with the PNIPAM aqueous solution, a similar trend of OH groups can be observed during neat water diffusion where a striking increase takes place at the second step. It seems that water molecules can also diffuse spontaneously into the IL phase without the aid of PNIPAM because of elevated solubility at high temperature.56,57 However, for neat water diffusion in Fig. 5b, further interpretation related to the hysteresis of rapid diffusion of water and higher equilibrium concentration of water is still unclear. On the one hand, it is well known that many water⋯amide (PNIPAM) hydrogen bonds survive around the aggregations of PNIPAM even above LCST,32–34,58 also confirmed by variable-temperature FTIR in Fig. S2 (ESI†). It is thus reasonable to believe that water molecules bundled with PNIPAM aggregations can transfer into IL phase together at the second stage, namely, this cooperation effect encourages some water molecules to diffuse in advance. On the other hand, very low interaction parameter59 and favourable interactions between PNIPAM and IL can infer that PNIPAM and hydrophobic IL are highly compatible at 80 °C. As a result, more hydrophobic PNIPAM segments can effectively hinder further diffusion of water into IL so that there is a lower water concentration in PNIPAM/IL layer in contrast with neat water diffusion (Fig. 5b). In other words, residual hydrated groups of PNIPAM induce the earlier diffusion of water with aid of the cooperation effect, but reduced hydrophobicity of PNIPAM prevents other water molecules from diffusing into the IL layer once PNIPAM chains are dissolved sufficiently in [C2MIM][NTf2].
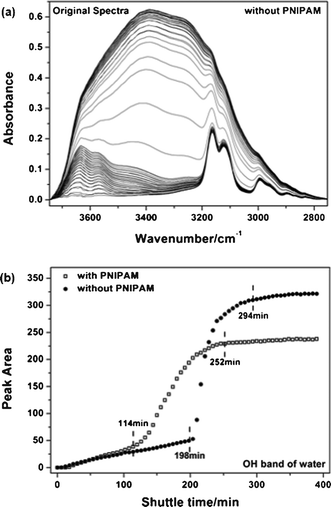 |
| | Fig. 5 (a) Original spectra in the region (3745–2751 cm−1) at intervals of 6 min during neat water diffusion under the same conditions as the PNIPAM transfer system. (b) Comparative changes of the OH band intensity as a function of diffusion time in the presence or absence of PNIPAM. | |
The structural changes of ternary components
To further understand the structures of PNIPAM transfer system, comparative analysis is processed in detail based on the bending vibration of NH bands (δNH), stretching vibration of CH3 bands (νCH3) and stretching vibration of OH bands (νOH).
δ
NH and νCH3 bands of PNIPAM.
Fig. 6a and b show the original and second derivative spectra of δNH in PNIPAM transfer system and 10% PNIPAM/[C2MIM][NTf2] system. The band positions are listed in Table 1 where it can be found that three bands only appear below the UCST in the 10% PNIPAM/[C2MIM][NTf2] system. Previously, we observed that an opaque PNIPAM gel was observed below the UCST due to the increase of additional NH⋯OC amide hydrogen bonds which led to the aggregation formed in the IL.37 Therefore, the 1546 cm−1 band can be assigned to NH⋯OC hydrogen bonds while the other two bands at 1537 and 1512 cm−1 are attributed to NH⋯anion of IL and free NH. It is the interaction of NH⋯anion that induces the PNIPAM chains to unfold in the IL solution above the UCST. However, a blue shift to 1541 cm−1 can be noticed clearly in the PNIPAM transfer system, which is most likely to be from the influence of water hydrogen bands, namely, complex interactions (water⋯NH⋯anion) between NH bands, H2O and anion of IL. Similarly, the band of “free” NH is also affected by water diffusion due to the higher wavenumber at 1521 cm−1. Fig. 6c shows that hydrated CH3 bands (2979 cm−1) also undergo a blue shift in the PNIPAM transfer system compared with the 10% PNIPAM/IL system (2974 cm−1) although the level of hydration is not as high as that in the 10% PNIPAM/D2O system (2982 cm−1). Generally, PNIPAM molecules will form some complex structures with water and IL through both hydrogen bonds and interaction of electric charges (water⋯PNIPAM⋯anion) when water as well as PNIPAM diffuses into the hydrophobic IL.
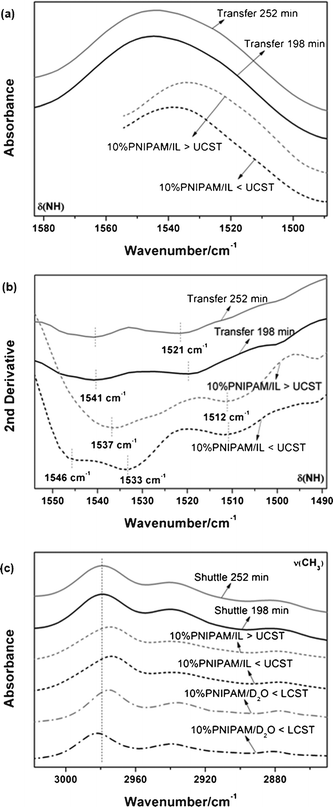 |
| | Fig. 6 (a) Original spectra of the δNH band in the PNIPAM transfer system (solid lines) and in the 10% PNIPAM/IL system (dashed lines) obtained at different temperatures (above and below the UCST). (b) Second derivatives of δNH band based on the original spectra. (c) Original spectra of νCH3 band in the PNIPAM transfer system, the 10% PNIPAM/IL system and the 10% PNIPAM/D2O system (dash-dot lines). | |
Table 1 FTIR wavenumbers of δNH and νCH3 bands in experimental conditions of different systems
| System |
Experimental conditions |
Wavenumber/cm−1 |
|
Wavenumber cannot be measured in the second derivative.
Values not obtained due to deuteration.
|
| Transfer with PNIPAM |
252 min |
—a |
1541 |
1521 |
2979 |
| 198 min |
—a |
1541 |
1520 |
2979 |
| |
| 10% PNIPAM/IL |
T > UCST |
—a |
1537 |
1512 |
2974 |
|
T < UCST |
1546 |
1533 |
1512 |
2974 |
| |
| 10% PNIPAM/D2O |
T > LCST |
—b |
—b |
—b |
2975 |
|
T < LCST |
—b |
—b |
—b |
2982 |
ν
OH band of water.
There are several spectra of OH bands at some important transfer points in Fig. 7a which reveals the pronounced changes of OH bands as a function of transfer time. Table 2 describes the band positions at different transfer times according to the results of second derivative spectra (Fig. 7b). At the initial transfer (24 min), only two narrow bands at 3644 and 3566 cm−1 can be observed, which are assigned to the antisymmetric and symmetric stretching vibrations of water monomer forming symmetric 1:2-type hydrogen-bonded complexes (anion⋯H2O⋯anion), in accord with many previous reports.45,60–63 This infers that the monomer structure can be formed when only a small number of water molecules diffuse into the IL phase. The OH region (3500–3300 cm−1) is enhanced slightly at 114 min in the conventional spectra but the change is too small to observe two split peaks in the second derivative, and so monomer structures still make up the vast majority in the IL phase before 114 min. At 156 min, the middle time of the second stage where water and PNIPAM molecules diffuse into IL together in the form of H2O⋯PNIPAM aggregations discussed above, it is observed that bands at 3431, 3389 and 3295 cm−1 increase considerably. The diffusion generates more hydrogen bonded water molecules and water clusters such as (H2O)n⋯PNIPAM⋯anion and anion⋯(H2O)n⋯anion (n ≥ 2). After 198 min when PNIPAM molecules have completed their transfer, the OH region still continues to grow substantially, especially in the range of 3500–3300 cm−1. This indicates that more and more water cluster structures are formed due to further diffusion, up to 252 min, the end of the third step. Moreover, some OH bands exhibit significant red shifts during the transfer process (3644 cm−1 at 24 min shifting to 3635 cm−1), with the 3566 cm−1 band almost lost after 198 min, both of which can be rationalized by the original water structure (anion⋯H2O⋯anion) being replaced by (H2O)n⋯PNIPAM⋯anion chains and anion⋯(H2O)n⋯anion cluster units.
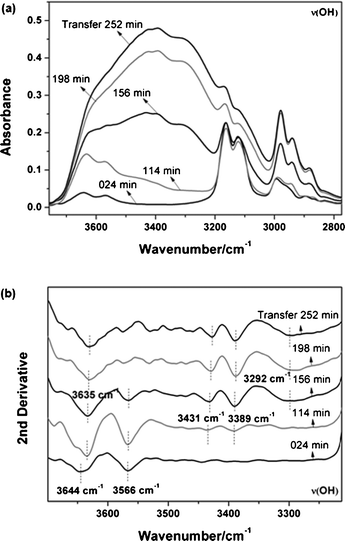 |
| | Fig. 7 (a) Original spectra of the νOH band in the PNIPAM transfer system at different transfer times (252, 198, 156, 114, 24 min). (b) Second derivatives of the νOH band based on their original spectra. | |
Table 2 FTIR wavenumbers of the νOH band at different times of the PNIPAM transfer system
| System |
Experimental conditions |
Wavenumber/cm−1 |
|
Wavenumber cannot be measured in the second derivative; (w) indicates the bands are very weak.
|
| Transfer with PNIPAM |
252 min |
3635 |
—a |
3431 |
3389 |
3292 |
| 198 min |
3635 |
—a |
3431 |
3389 |
3292 |
| 156 min |
3635 |
3566 |
3431 |
3389 |
3292 |
| 114 min |
3635 |
3566 |
3435 (w) |
3390 (w) |
—a |
| 24 min |
3644 |
3566 |
—a |
—a |
—a |
As result of the complex interactions among ternary components, a variety of structures are obtained with PNIPAM and water diffusion in the transfer system, as summarized in Scheme 2. When only a small number of water molecules start to diffuse in the first step, the main structure in the IL phase is a symmetric 1:2-type hydrogen bonded form: anion⋯H2O⋯anion between IL and water. Then both water and PNIPAM transfer quickly during the second phase. In that time, some complex interactions can be observed such as H2O⋯PNIPAM⋯anion and anion⋯(H2O)n⋯anion. In the third step from 198 to 252 min, water molecules continue to diffuse rapidly after the end of PNIPAM transfer, and more and more water clusters (H2O)n⋯PNIPAM⋯anion and anion⋯(H2O)n⋯anion can be formed in the ternary system. After 252 min, all of components tend to level off in the IL phase.
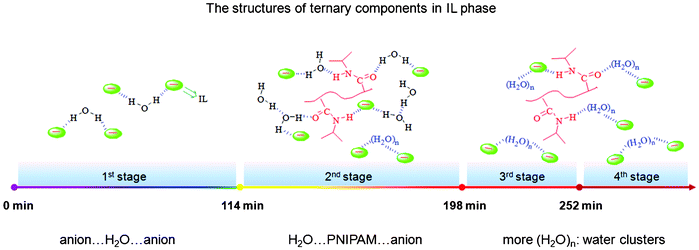 |
| | Scheme 2 The schematic structures of ternary components in the hydrophobic IL phase at different transfer stages. | |
Conclusion
This transfer system includes not only PNIPAM and water diffusion but also some significant interactions among ternary components. The whole dynamic process can be divided into four steps according to in situ infrared spectroscopy and MCR. Actually, due to favorable interactions between PNIPAM and IL at high temperature, PNIPAM transfer only takes place in the second step where water molecules bonded with PNIPAM aggregation together transfer into IL phase. This cooperation effect induces earlier diffusion of water into the hydrophobic IL compared with the neat water diffusion system. On the other hand, however, more hydrophobic PNIPAM segments can hinder further diffusion of water into [C2MIM][NTf2] due to high compatibility between PNIPAM and IL at high temperature. In this period, some complex interactions among ternary components can be observed in the PNIPAM/H2O transfer system such as H2O⋯PNIPAM⋯anion and anion⋯(H2O)n⋯anion, in contrast with other binary systems. However, it is found that water diffusion lasts for a much longer time than PNIPAM. A small number of water molecules first diffuse into IL at the initial stage where a symmetric hydrogen-bonded form: anion⋯H2O⋯anion can be clearly detected from two narrow OH bands ranging from 3700 to 3500 cm−1. In the third stage, water continues to diffuse and forms more and more water clusters with PNIPAM and IL although polymer transfer is completed. Finally, all of components (PNIPAM, water and IL) are stabilized in the IL phase by optimization of complementary and complex interactions.
Acknowledgements
We gratefully acknowledge the financial support from National Science Foundation of China (NSFC) (20934002), and the National Basic Research Program of China (No. 2009CB930000).
References
- H. Yang, R. S. Cheng and Z. L. Wang, Polymer, 2003, 44, 7175–7180 CrossRef CAS.
- H. W. Chen, J. F. Li, Y. W. Ding, G. Z. Zhang, Q. J. Zhang and C. Wu, Macromolecules, 2005, 38, 4403–4408 CrossRef CAS.
- Y. J. Lu, K. J. Zhou, Y. W. Ding, G. Z. Zhang and C. Wu, Phys. Chem. Chem. Phys., 2010, 12, 3188–3194 RSC.
- H. J. Lai, G. T. Chen, P. Y. Wu and Z. C. Li, Soft Matter, 2012, 8, 2662–2670 RSC.
- X. M. Lian, J. Jin, J. Tian and H. Y. Zhao, ACS Appl. Mater. Interfaces, 2010, 2, 2261–2268 CAS.
- A. Berteotti, A. Barducci and M. Parrinello, J. Am. Chem. Soc., 2011, 133, 17200–17206 CrossRef CAS.
- R. Kumaran and P. Ramamurthy, J. Fluoresc., 2011, 21, 1499–1508 CrossRef CAS.
- H. B. Gray, J. R. Winkler and J. J. Kozak, Mol. Phys., 2012, 110, 419–429 CrossRef CAS.
- K. L. Deng, H. B. Zhong, X. Y. Zheng, X. H. Song, H. Tian, P. F. Zhang, X. B. Ren and H. J. Wang, Polym. Adv. Technol., 2010, 21, 584–590 CAS.
- R. Regmi, S. R. Bhattarai, C. Sudakar, A. S. Wani, R. Cunningham, P. P. Vaishnava, R. Naik, D. Oupicky and G. Lawes, J. Mater. Chem., 2010, 20, 6158–6163 RSC.
- C. Chang, H. Wei, D. Q. Wu, B. Yang, N. Chen, S. X. Cheng, X. Z. Zhang and R. X. Zhuo, Int. J. Pharm., 2011, 420, 333–340 CrossRef CAS.
- J. Ye, J. Xu, J. M. Hu, X. F. Wang, G. Z. Zhang, S. Y. Liu and C. Wu, Macromolecules, 2008, 41, 4416–4422 CrossRef CAS.
- M. Li, P. De, H. M. Li and B. S. Sumerlin, Polym. Chem., 2010, 1, 854–859 RSC.
- J. Ramos, A. Imaz and J. Forcada, Polym. Chem., 2012, 3, 852–856 RSC.
- H. Wei, S. X. Cheng, X. Z. Zhang and R. X. Zhuo, Prog. Polym. Sci., 2009, 34, 893–910 CrossRef CAS.
- R. Pamies, K. Z. Zhu, A. L. Kjoniksen and B. Nystrom, Polym. Bull., 2008, 62, 487–502 CrossRef.
- H. Q. Bao, L. Li, W. C. Leong and L. H. Gan, J. Phys. Chem. B, 2010, 114, 10666–10673 CrossRef CAS.
- Y. Xia, X. C. Yin, N. A. D. Burke and H. D. H. Stover, Macromolecules, 2005, 38, 5937–5943 CrossRef CAS.
- X. M. Lian, D. X. Wu, X. H. Song and H. Y. Zhao, Macromolecules, 2010, 43, 7434–7445 CrossRef CAS.
- J. Xu, J. Ye and S. Y. Liu, Macromolecules, 2007, 40, 9103–9110 CrossRef CAS.
- I. Shechter, O. Ramon, I. Portnaya, Y. Paz and Y. D. Livney, Macromolecules, 2010, 43, 480–487 CrossRef CAS.
- Z. G. Gao, X. D. Tao, Y. Cui, T. Satoh, T. Kakuchi and Q. Duan, Polym. Chem., 2011, 2, 2590–2596 RSC.
- P. M. Reddy and P. Venkatesu, J. Phys. Chem. B, 2011, 115, 4752–4757 CrossRef CAS.
- J. Yip, J. Duhamel, X. P. Qiu and F. M. Winnik, Macromolecules, 2011, 44, 5363–5372 CrossRef CAS.
- H. Inoue, K. Katayama, K. Iwai, A. Miura and H. Masuhara, Phys. Chem. Chem. Phys., 2012, 14, 5620–5627 RSC.
- X. H. Wang and C. Wu, Macromolecules, 1999, 32, 4299–4301 CrossRef CAS.
- S. Mendrek, A. Mendrek, H. J. Adler, A. Dworak and D. Kuckling, Colloid Polym. Sci., 2010, 288, 777–786 CAS.
- C. Zhou, M. A. Hillmyer and T. P. Lodge, Macromolecules, 2011, 44, 1635–1641 CrossRef CAS.
- L. Starovoytova, J. Spevacek and M. Ilavsky, Polymer, 2005, 46, 677–683 CrossRef CAS.
- C. M. Burba, S. M. Carter, K. J. Meyer and C. V. Rice, J. Phys. Chem. B, 2008, 112, 10399–10404 CrossRef CAS.
- J. Q. Chen, X. L. Gong, H. Yang, Y. F. Yao, M. Xu, Q. Chen and R. S. Cheng, Macromolecules, 2011, 44, 6227–6231 CrossRef CAS.
- Y. Maeda, T. Higuchi and I. Ikeda, Langmuir, 2000, 16, 7503–7509 CrossRef CAS.
- Y. Maeda, T. Nakamura and I. Ikeda, Macromolecules, 2001, 34, 8246–8251 CrossRef CAS.
- B. Sun, Y. Lin, P. Wu and H. W. Siesler, Macromolecules, 2008, 41, 1512–1520 CrossRef CAS.
- H. Cheng, L. Shen and C. Wu, Macromolecules, 2006, 39, 2325–2329 CrossRef CAS.
- T. Ueki and M. Watanabe, Chem. Lett., 2006, 35, 964–965 CrossRef CAS.
- Z. W. Wang and P. Y. Wu, J. Phys. Chem. B, 2011, 115, 10604–10614 CrossRef CAS.
- Z. F. Bai, Y. Y. He, N. P. Young and T. P. Lodge, Macromolecules, 2008, 41, 6615–6617 CrossRef CAS.
- Z. F. Bai and T. P. Lodge, J. Phys. Chem. B, 2009, 113, 14151–14157 CrossRef CAS.
- Z. F. Bai and T. P. Lodge, Langmuir, 2010, 26, 8887–8892 CrossRef CAS.
- C. Guerrero-Sanchez, D. Wouters, S. Hoeppener, R. Hoogenboom and U. S. Schubert, Soft Matter, 2011, 7, 3827–3831 RSC.
- Y. Y. He and T. P. Lodge, J. Am. Chem. Soc., 2006, 128, 12666–12667 CrossRef CAS.
- Z. F. Bai, Y. Y. He and T. P. Lodge, Langmuir, 2008, 24, 5284–5290 CrossRef CAS.
- C. Guerrero-Sanchez, J. F. Gohy, C. D'Haese, H. Thijs, R. Hoogenboom and U. S. Schubert, Chem. Commun., 2008, 2753–2755 RSC.
- L. Cammarata, S. G. Kazarian, P. A. Salter and T. Welton, Phys. Chem. Chem. Phys., 2001, 3, 5192–5200 RSC.
- L. P. Zhang, I. Noda and Y. Q. Wu, Appl. Spectrosc., 2009, 63, 694–699 CrossRef CAS.
- Y. Maeda, H. Yamauchi, M. Fujisawa, S. Sugihara, I. Ikeda and S. Aoshima, Langmuir, 2007, 23, 6561–6566 CrossRef CAS.
- Y. Jin, W. Wang and Z. Su, Macromolecules, 2011, 44, 2132–2139 CrossRef CAS.
- S. Morita, M. Tanaka and Y. Ozaki, Langmuir, 2007, 23, 3750–3761 CrossRef CAS.
- L. S. Wan, X. J. Huang and Z. K. Xu, J. Phys. Chem. B, 2007, 111, 922–928 CrossRef CAS.
- D. T. Hallinan and Y. A. Elabd, J. Phys. Chem. B, 2009, 113, 4257–4266 CrossRef CAS.
- J. M. Andanson, F. Jutz and A. Baiker, J. Phys. Chem. B, 2010, 114, 2111–2117 CrossRef CAS.
- J. M. Andanson, F. Jutz and A. Baiker, J. Supercrit. Fluids, 2010, 55, 395–400 CrossRef CAS.
- F. J. Deive, A. Rodriguez, A. B. Pereiro, J. M. M. Araujo, M. A. Longo, M. A. Z. Coelho, J. N. C. Lopes, J. Esperanca, L. P. N. Rebelo and I. M. Marrucho, Green Chem., 2011, 13, 390–396 RSC.
- S. Sasic, Y. Ozaki, A. Olinga and H. W. Siesler, Anal. Chim. Acta, 2002, 452, 265–276 CrossRef CAS.
- M. G. Freire, C. Neves, P. J. Carvalho, R. L. Gardas, A. M. Fernandes, I. M. Marrucho, L. Santos and J. A. P. Coutinho, J. Phys. Chem. B, 2007, 111, 13082–13089 CrossRef CAS.
- C. Neves, M. L. S. Batista, A. F. M. Claudio, L. Santos, I. M. Marrucho, M. G. Freire and J. A. P. Coutinho, J. Chem. Eng. Data, 2010, 55, 5065–5073 CrossRef CAS.
- Y. Ono and T. Shikata, J. Phys. Chem. B, 2007, 111, 1511–1513 CrossRef CAS.
- C. Soto-Figueroa, M. D. Rodriguez-Hidalgo and L. Vicente, Soft Matter, 2012, 8, 1871–1877 RSC.
- Y. Wang, H. R. Li and S. J. Han, J. Phys. Chem. B, 2006, 110, 24646–24651 CrossRef CAS.
- Y. Jeon, J. Sung, D. Kim, C. Seo, H. Cheong, Y. Ouchi, R. Wawa and H. O. Hamaguchi, J. Phys. Chem. B, 2008, 112, 923–928 CrossRef CAS.
- B. J. Sun and P. Y. Wu, J. Phys. Chem. B, 2010, 114, 9209–9219 CrossRef CAS.
- J. M. Andanson, M. J. Beier and A. Baiker, J. Phys. Chem. Lett., 2011, 2, 2959–2964 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ra21288a |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.