Production of high quality fuels from lignocellulose-derived chemicals: a convenient C–C bond formation of furfural, 5-methylfurfural and aromatic aldehyde†
Yao-Bing
Huang
,
Zhen
Yang
,
Jian-Jun
Dai
,
Qing-Xiang
Guo
and
Yao
Fu
*
Department of Chemistry, Anhui Province Key Laboratory of Biomass Clean Energy, University of Science and Technology of China, Hefei 230026. E-mail: fuyao@ustc.edu.cn
First published on 19th September 2012
Abstract
A new route of converting the whole of lignocellulose biomass derived molecules into high quality fuels was developed with C–C bond formation as the key step. The fuel precursors with 10–14 carbons could be easily obtained though direct self-coupling of furfural, 5-methyfurfural or aromatic aldehyde, the main components of lignocellulose, under mild conditions in water. After dehydration/hydrogenation, straight or branched alkanes with 8–14 carbons within diesel ranged fuels were obtained in moderate to high yield.
Current huge consumption of fossil resources and the resulting CO2 emission is forcing mankind to develop benign and unlimited energy systems based on renewable resources.1 Among the promoted forms of new energy sources, biomass is considered as an ideal substitute for the limited fossil resources, because it is the only sustainable source of organic carbon available on earth.2 The first generation of biofuels mainly included bioethanol and biodiesel, but their production requires crop land and competes with food production. Alternatively, a second generation of biofuels from lignocellulose biomass has been proposed. The lignocellulose mostly comes from plants and wood, which are more abundant and have a low cost.3 It consists of three primary components, cellulose, hemicelluloses and lignin. These biopolymers need to be depolymerized and further upgraded to biofuels.4 Moreover, the hexose, pentose and aromatic units with 5–6 carbons after depolymerization have to be condensed though C–C bond formation to obtain C8–C15 intermediates for the diesel-range fuel fraction. Thus, it is essential to develop synthetic methods for the efficient transformation of the whole lignocellulose to high quality biofuels.
Currently, there are several routes to converting the biomass-derived carbohydrates into fuels, such as fast pyrolysis and gasification, and Fischer–Tropsch synthesis.5,6 Another simple route starting from lignocelluloses has attracted much attention and it involves: (1) depolymerization of the biopolymer into monomers and subsequent conversion into platform molecules like hydroxymethyl-furfural (HMF), furfural or vanillin, followed by (2) C–C bond formation and hydrodeoxygenation of the molecules to obtain C8–C15 carbon alkanes within the diesel fuels. Whereas important progress has been achieved for the production of platform molecules in the first step,7 studies on C–C bond formation of these molecules in the second step remain rare and need further development.
To produce larger fuel precursors (8–15 carbons) from carbohydrate derived C5–C6 molecules, Dumesic et al. reported pioneering work on the conversion of biomass-derived carbohydrates into liquid alkanes by aqueous processing. A new C–C bond between HMF (or furfural) and acetone formed through aldol condensation, resulting in a C13–C15 molecule fraction.8 Another approach involves decarboxylation of γ-valerolactone (GVL) into a gas stream containing butane and CO2. Then the stream forms new C–C bonds to produce long chain alkanes at high temperatures and pressures.9 Besides, GVL can undergo a ring-opening reaction, leading to the intermediate of pentanoic acid that can subsequently form 5-nonanone by a ketonization reaction.10 In addition, levulinic acid can be converted to octane by a two-step electrochemical conversion via pentanoic acid.11 Recently, Corma et al. reported a process of converting 2-methylfuran (2MF) into high-quality diesel by acid catalyzed condensation and hydrodeoxygenation.12 Thayumanavan and Huber et al. showed several C–C bond formation reactions of converting biomass-derived molecules into large species, which may find applications in the conversion of biomass into diesel fuel (Scheme 1).13
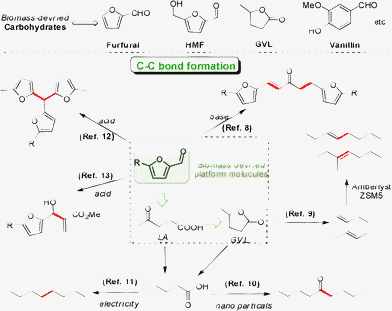 | ||
| Scheme 1 Previous work on C–C bond formation of biomass-derived molecules. | ||
Inspired by these previous findings, we were interested in the exploration of potential chemical transformation routes for constructing new C–C bonds between the C5–C6 platform molecules. To the best of our knowledge, there is no straight example of producing fuel precursors from the platform molecule by a one-step reaction. Herein, we present our investigation on converting lignocellulose-derived platform molecules to liquid alkane fuels (Scheme 2). Three types of molecules (furfural, 5-methylfurfural and aromatic aldehydes) derived from cellulose, hemicellulose, and lignin can effectively undergo self-coupling by using reductants at room temperature under atmosphere. Moreover, the reaction could be conducted in water, which is considered to be a green medium for chemical synthesis. The new route not only provides a new type of C–C bond forming reaction for platform molecules in concept, but also shows an additional potential route for converting all the lignocellulose components into liquid transportation fuels.
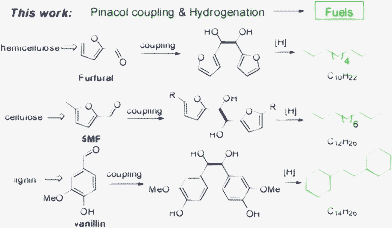 | ||
| Scheme 2 Our route to construct a new C–C bond. | ||
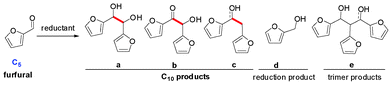
We begin our study by choosing furfural as a model substance to explore the reaction conditions for C–C bond formation. Furfural, derived from hemicelluloses, is produced on a large scale in industry.14 It contains a formyl group on the α-position, which may be a reactive site for constructing a new C–C bond. During the test, 1,2-diols with two furan rings are formed. According to the previous work, aryl aldehydes underwent pinacolic coupling via the radical reaction pathway under some reductive system (Scheme 3).15 Thus, various metal reagents were used to optimize the reaction conditions. Our previous work on reductive coupling revealed that a catalytic amount of bromobenzene with alkali can mediate the coupling with a high yield at room temperature,16 but the result turned out to be unsatisfactory in our test (Table 1, entry 1). Strikingly, a mixture of products were obtained when TiCl3, SmI2 and VCl3 were used with different metals (Table 1, entries 2–6). Among these products, a was the self-coupling product of furfural. It was composed of dl-, meso-diastereoisomers, which could be explained by the different intermediates formed in Scheme 3. b and c were the products after removal of a molecule of H2 or H2O from a. d and e were the reductive or trimer products of furfural (see ESI†). Note that a, b and c were all the precursors of C10 alkanes; they could be classified as C10 products. To our delight, a good yield (88% conversion, 87% selectivity) was achieved when metallic Al powder was used in 10% NaOH solution (Table 1, entry 7). An excess of reductant (2 equivalents) led to the side product (furfuryl alcohol) in 29% yield (Table 1, entry 8). With the medium changed to 10% aq. KOH, the reaction achieved a maximum conversion of 99%, but the selectivity of a dropped to 49% while that of c increased to 40% (Table 1, entry 9). For Mn and Mg, the best reaction medium was 0.1 M NH4Cl, and an excess of the reductant was necessary (Table 1, entries 10–13). When the reductant was changed to Zn, the conversion was 93% and the C10 selectivity was as high as 98% in 6 h (Table 1, entry 14). Excess reductant reduced the C10 selectivity to 89% with 10% furfural alcohol as the reduction product (Table 1, entry 15). The reaction could even finish in 1 h (Table 1, entry 16). In addition, the good performances of the reaction under 0 °C or 50 °C indicated that the proposed protocol was easy to operate under a wide range of temperatures (Table 1, entries 18–19). The resulting metallic oxide generated from the metals in the solution could be collected and smelted to zero-valence metals in the smelter for further recycling. Overall, the C–C bond formation between two furfural molecules could be achieved with a high yield of C10 product by using cheap metals in aqueous solution under mild conditions.
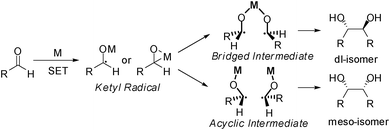 | ||
| Scheme 3 The mechanism for the reductive coupling of furfural. | ||
| Run | Reductant | Molar equiv. | Reaction mediumb | Conv.b(%) | Selectivityb (%) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| a/b/c | C10 | d | e | |||||||
| a Conditions: furfural 2.0 mmol, solvents 3 ml, room temperature, 6 h. b Conversion and selectivity were based on GC-MS. c TiCl3 in 15% acidic aqueous solution. d 0.1 mol L−1 SmI2 in THF. e Reaction for 1 h. f Reaction at 0 °C. g Reaction at 50 °C. | ||||||||||
| 1 | Li/PhBr | 1/0.1 | neat | N. R | —/—/— | — | — | — | ||
| 2c | TiCl3 | 1 | H2O | N. R | —/—/— | — | — | — | ||
| 3c | TiCl3/Zn | 1/1 | H2O | 50 | 92/6/— | 98 | 3 | 1 | ||
| 4d | SmI2/Zn | 0.1/1 | H2O | 20 | 80/10/— | 90 | 5 | — | ||
| 5d | SmI2/Mg | 0.1/1 | H2O | 24 | 91/—/— | 91 | 2 | — | ||
| 6 | VCl3/Al | 1/1 | H2O | 70 | 95/1/— | 96 | — | — | ||
| 7 | Al | 1 | 10% NaOH | 88 | 87/4/— | 91 | 8 | — | ||
| 8 | Al | 2 | 10% NaOH | 92 | 62/7/— | 69 | 29 | — | ||
| 9 | Al | 2 | 10% KOH | 99 | 49/2/40 | 91 | 7 | — | ||
| 10 | Mn | 1 | 0.1 M NH4Cl | 2 | —/—/— | — | — | — | ||
| 11 | Mn | 3 | 0.1 M NH4Cl | 59 | 52/—/— | 52 | 3 | 41 | ||
| 12 | Mg | 1 | 0.1 M NH4Cl | N. R. | —/—/— | — | — | — | ||
| 13 | Mg | 3 | 0.1 M NH4Cl | 84 | 89/7/1 | 97 | 3 | — | ||
| 14 | Zn | 1 | 10% NaOH | 93 | 93/5/— | 98 | 1 | — | ||
| 15 | Zn | 2 | 10% NaOH | 95 | 85/4/— | 89 | 10 | 1 | ||
| 16e | Zn | 1 | 10% NaOH | 92 | 89/6/— | 95 | 4 | — | ||
| 17 | Zn | 1 | 5% NaOH | N. R | —/—/— | — | — | — | ||
| 18f | Zn | 1 | 10% NaOH | 94 | 88/6/1 | 95 | 4 | 1 | ||
| 19g | Zn | 1 | 10% NaOH | 99 | 96/—/— | 96 | — | 1 | ||
With the optimized conditions, we turned to the C–C bond formation of HMF or 5-methylfurfual (5MF) in Scheme 4. Indeed, HMF or 5MF could be obtained from fructose or cellulose through a one-step procedure.7,17 Besides, C6 sugars are more versatile and available than C5 sugars in nature, so that it is essential to develop a progress by which diesel-ranged fuels can be produced from HMF or 5MF. By applying the optimized conditions to HMF, a mixture of products with molecule weights ranging from 200 to 800 was detected, primarily the polymers formed between HMF. The result may be ascribed to the low stability of HMF under the reaction conditions, which often hampered its application. In another method, 5MF was a proper substance, the synthesis of the C12 fuel precursor was carried out under optimized conditions and a dimmer product was obtained with a high yield up to 85%.
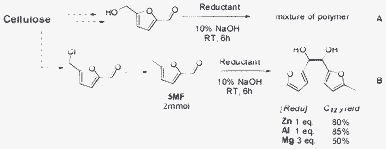 | ||
| Scheme 4 Reductive coupling of cellulose-derived molecules (A) HMF (B) 5MF. | ||
As we know, lignin accounts for about 15–30% of lignocellulose by weight and 40% by energy. Many studies18 have concentrated on the conversion of lignin and lignin-derived phenols to various aromatic chemicals and fuels. Kou, Lercher and Dyson etc. reported the effective conversion of lignin-based phenols or bio-oil into alkanes with heterogeneous catalysts in acidic aqueous solution or ionic liquid.19 However, these monomer phenols always contain 6–9 carbon atoms, which makes it difficult to meet the requirements of diesel fuels, which range from C8 to C15. Alternatively, aromatic aldehydes, such as vaillian and veratraldehyde, could be selectively obtained from lignin through chemical transformations.20 They could be the reactants for C–C bond formation. Under our conditions, various lignin-derived aromatic aldehydes could be converted to a C14 fuel precursor.
By using Mg powder in acidic aqueous solution, dimer products A were obtained in a moderate to high yield (Scheme 5). Some 1,2-diol products could be converted to B though pinacol rearrangement under acidic conditions (see Scheme S1, ESI†).
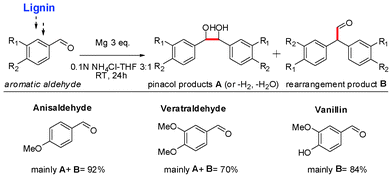 | ||
| Scheme 5 Reductive coupling of lignin derived molecules. | ||
After separation of these fuel precursors (dimer products), we turn to the second step for dehydration/hydrogenation of these fuel precursors into alkanes. In order to give straightforward examples, the dimer products obtained from furfural, 5-methylfurfural and anisaldehyde were chosen as the model fuel precursor, corresponding to C10, C12 and C14 alkanes. Noble metal nanoparticles supported on different carriers, such as Pt/NbOPO4, Pd/C, have proven to be effective and recyclable catalysts in the dehydration/hydrogenation process in the literature.8,12,21 Commercially available catalysts combined with solid acids were tested in our research. The results in Table 2 showed that Pt/C was more effective than Pd/C in removing the oxygen atoms in the substance, when combined with NbOPO4 in aqueous solution (Table 2, entries 1–2); the yield of C8 to C10 alkanes was up to 75.2%. Changing the solid acid to TaOPO4 and WO3 resulted in yields of 83.6% and 74.1%, respectively. Furthermore, the C12 and C14 alkanes could be obtained though hydrodeoxygenation of fuel precursors B and C (Table 2, entries 6–10). In order to avoid energy-costly separation of the pure fuel precursors from the reaction system, the reaction mixture was extracted and the organic layer was condensed to obtain the crude products A1–C1 (a mixture of raw materials, dimer products and some side products). These crude products were subsequently hydrodeoxygenated and the C8–C10 alkane yield was 70.6% (calculated from furfural). During the hydrodeoxygenation of these C10–C14 precursors, there were fewer C–C cleavage products (Cn−x, x = 1–3), oxygenated compounds, short-chain alkanes and some gaseous products (CO2, methane etc.) (see ESI†). However, these cleavage products with more than 8 carbon atoms can all be classified as fuels. Overall, the Pt/C catalysts combined with different solid acids were proven to effective catalysts in the dehydration/hydrogenation progress, and the fuel precursors were successfully transformed into alkanes with 8–14 carbons in high yield.
| Run | Feed | Cat. | Acid | Yield of products in the organic phase | ||||
|---|---|---|---|---|---|---|---|---|
| C8–C10 | C11–C12 | C13–C14 | Alkanes | [oxy]c | ||||
| a Conditions: dimer product 1.0 mmol, Pt/C (5 wt%), 100 mg, solid acids 50 mg, H2O 10 ml, 4 MPa H2, 573 K, 3 h. b The alkane yield from crude products was calculated from the aldehyde. The yield was determined by GC-MS using tridecane as internal standard and other side products (low-weight hydrocarbons, some gaseous products) were omitted in the final calculation. c Mixture of oxygen compounds. | ||||||||
| 1 | A | Pd/C | NbOPO4 | 26 | — | — | 26 | 70.4 |
| 2 | Pt/AC | NbOPO4 | 75.2 | — | — | 75.2 | 16.2 | |
| 3 | TaOPO4 | 83.6 | — | — | 83.6 | 9.4 | ||
| 4 | WO3 | 74.1 | — | — | 74.1 | 13.5 | ||
| 5b | A 1 | TaOPO4 | 76.5 | — | — | 76.5 | 8.6 | |
| 6 | B | Pt/AC | NbOPO4 | 6.3 | 79.4 | — | 85.7 | 5.2 |
| 7 | B | TaOPO4 | 30.4 | 52.0 | — | 82.4 | 3.9 | |
| 8b | B 1 | NbOPO4 | 4.3 | 67.8 | — | 72.1 | 3.7 | |
| 9 | C | Pt/AC | TaOPO4 | — | — | 91.5 | 91.5 | 2.5 |
| 10b | C 1 | TaOPO4 | — | — | 83.2 | 83.2 | 2.0 | |
In summary, the present study demonstrates the possibility of converting the whole of lignocellulose component derived molecules (furfural, 5-methylfurfural and aromatic aldehydes) into high quality fuels. The first step of self-condensation could be easily carried out with cheap metal powders at ambient temperature in water. With simple extraction, the hydroxyalkylation step could be achieved by using commercial Pt/C combined with solid acids in water. The C10, C12 and C14 alkanes can be selectivity obtained with a high yield up to 83%. The route not only provides a conceptual new C–C bond formation reaction of producing fuel precursors directly from biomass derived platform molecules, but also shows an additional example of converting lignocelluse-derived C5–C6 molecules to diesel range fuels. However, there were still unavoidable limitations with the above route, and more economic and green electron donor systems need to be developed. Research based on the production of diesel range fuels from biomass is a complex and systematic work, so interdisciplinary cooperation and exploration are necessary. The present explorative route was just part of it. We are expecting that the potential route presented here would be a inspiration for synthetic chemists and chemical engineers devoted to the scientific research of biomass conversion in the laboratory or in the refining industry.
Acknowledgements
We thank the 973 Program (2012CB215305, 2012CB215306), NSFC (21172209) and CAS (KJCX2-EW-J02) for the financial support.References
- (a) T. E. Bull, Science, 1999, 285, 1209 CrossRef CAS; (b) A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick Jr., J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484 CrossRef CAS.
- (a) D. L. Klass, Biomass for the Renewable Energy, Fuels and Chemicals, Academic press, London, 1998 Search PubMed; (b) J.-P. Lange, E. Heide, J. Buijtenen and R. Price, ChemSusChem, 2012, 5, 150 CrossRef CAS.
- (a) C. Okkerse and H. van Bekkum, Green Chem., 1999, 1, 107 RSC; (b) B. E. Dale, S. Kim, Biomass refining global impact the biobased economy of the 21th century, in Biorefineries–Industrial Processes and Product, ed. B. Kamm, P. R. Gruber and M. Kamm, Wiley-VCH, Weinheim, 2006, vol. 1, pp. 41–66 Search PubMed.
- (a) E. L. Kunkes, D. A. Simonetti, R. M. West, J. C. Serrano-Ruiz, C. A. Gartner and J. A. Dumesic, Science, 2008, 322, 417 CrossRef CAS; (b) D. M. Alonso, J.Q. Bond and J.A. Dumesic, Green Chem., 2010, 12, 1493 RSC; (c) M. Mascal and E. B. Nikitin, Angew. Chem., Int. Ed., 2008, 47, 7924 CrossRef CAS; (d) J. B. Binder and R. T. Raines, J. Am. Chem. Soc., 2009, 131, 1979 CrossRef CAS; (e) G. W. Huber and A. Corma, Angew. Chem., Int. Ed., 2007, 46, 7184 CrossRef CAS; (f) J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164 CrossRef CAS; (g) J. C. Serrano-Ruiz and J. A. Dumesic, Energy Environ. Sci., 2011, 4, 83 RSC; (h) J. C. Serrano-Ruiz, R. Luque and A. Sepulveda-Escribano, Chem. Soc. Rev., 2011, 40, 5266 RSC; (i) F. M. A. Geilen, B. Engendahl, A. Harwardt, W. Marquardt, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2010, 49, 5510 CrossRef CAS.
- (a) O. O. James, S. Maity, L. A. Usman, K. O. Ajanaku, O. O. Ajani, T. O. Siyanbola, S. Sahu and R. Chaubey, Energy Environ. Sci., 2010, 3, 1833 RSC; (b) E. F. Iliopoulou, Curr. Org. Synth., 2010, 7, 587 CrossRef CAS.
- (a) A. V. Bridgwater and G. V. C. Peacocke, Renewable Sustainable Energy Rev., 2000, 4, 1 CrossRef CAS; (b) T. R. Carlson, T. P. Vispute and G. W. Huber, ChemSusChem, 2008, 1, 397 CrossRef CAS; (c) G.W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044 CrossRef CAS; (d) D. A. Bulusheva and J. R. H. Rossa, Catal. Today, 2011, 171, 1 CrossRef; (e) H. de Lasa, E. Salaices, J. Mazumder and R. Lucky, Chem. Rev., 2011, 111, 5404 CrossRef CAS.
- (a) A. Corma, S. Lborra and A. Velty, Chem. Rev., 2007, 107, 2411 CrossRef CAS; (b) H. B. Zhao, J. E. Holladay, H. Brown and Z. C. Zhang, Science, 2007, 316, 1597 CrossRef CAS; (c) J. B. Binder and R. T. Raines, J. Am. Chem. Soc., 2009, 131, 1979 CrossRef CAS; (d) A. A. Rosatella, S. P. Simeonov, R. F. M. Frade and C. A. M. Afonso, Green Chem., 2011, 13, 754 RSC; (e) G. Yong, Y. Zhang and J. Y. Ying, Angew. Chem., Int. Ed., 2008, 47, 9345 CrossRef CAS; (f) Pierre Gallezot, Chem. Soc. Rev., 2012, 41, 1538 RSC; (g) Y. Zhang and J. Y. G. Chan, Energy Environ. Sci., 2010, 3, 408 RSC.
- G. W. Huber, J. N. Chheda, C. J. Barrett and J. A. Dumesic, Science, 2005, 308, 1446 CrossRef CAS.
- J. Q. Bond, D. M. Alonso, D. Wang, R. M. West and J. A. Dumesic, Science, 2010, 327, 1110 CrossRef CAS.
- J. C. Serrano-Ruiz, D. Wang and J. A. Dumesic, Green Chem., 2010, 12, 574 RSC.
- P. Nilges, T. R. dos Santos, F. Harnisch and U. Schröder, Energy Environ. Sci., 2012, 5, 5231 CAS.
- A. Corma, O. de La Torre, M. Renz and N. Villandier, Angew. Chem. Int. Ed., 2011, 50, 2375 CAS.
- A. V. Subrahmanyam, S. Thayumanavan and G. W. Huber, ChemSusChem, 2010, 3, 1158 CrossRef CAS.
- (a) K. J. Zeitsch, The chemistry and technology of furfural and its many by-products, Elsevier, Amsterdam, 1st edn, 2000, vol. 13 Search PubMed; (b) R. H. Kottke, in Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, 2000, vol. 12, pp. 259–286 Search PubMed.
- (a) A. Gansauer and H. Bluhm, Chem. Rev., 2000, 100, 2771 CrossRef; (b) C.-J. Li, Chem. Rev., 2005, 105, 3095 CrossRef CAS; (c) C.-J. Li, Y. Meng and X.-H. Yi, J. Org. Chem., 1998, 63, 7498 CrossRef CAS.
- H. Zhao, D.-J. Li, L. Deng, L. Liu and Q.-X. Guo, Chem. Commun., 2003, 506 RSC.
- W. Yang and A. Sen, ChemSusChem, 2011, 4, 349 CrossRef CAS.
- (a) J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552 CrossRef CAS; (b) M. P. Pandey and C. S. Kim, Chem. Eng. Technol., 2011, 34, 29 CrossRef CAS; (c) F. G. Calvo-Flores and J. A. Dobado, ChemSusChem, 2010, 3, 1227 CrossRef CAS; (d) D.-Y. Hong, S. J. Miller, P. K. Agrawala and C. W. Jones, Chem. Commun., 2010, 46, 1038 RSC.
- (a) C. Zhao, Y. Kou, A. A. Lemonidou, X. Li and J. A. Lercher, Angew. Chem., Int. Ed., 2009, 48, 3987 CrossRef CAS; (b) N. Yan, Y. Yuan, R. Dykeman, Y. Kou and P. Dyson, Angew. Chem., Int. Ed., 2010, 49, 5549 CrossRef CAS; (c) N. Yan, C. Zhao, P. J. Dyson, C. Wang, L.-T. Liu and Y. Kou, ChemSusChem, 2008, 1, 626 CrossRef CAS.
- (a) M. B. Hocking, J. Chem. Educ., 1997, 74, 1055 CrossRef CAS; (b) M. Lersch, The Opportunities for Biorefineries, BIOREF-INTEG Workshop, Solihull, UK, 2009 Search PubMed; (c) E. A. Borges da Silva, M. Zabkova, J. D. Araffljo, C. A. Cateto, M. F. Barreiro, M. N. Belgacem and A. E. Rodrigues, Chem. Eng. Res. Des., 2009, 87, 1276 CrossRef CAS.
- W. Xu, Q. Xia, Y. Zhang, Y. Guo, Y. Wang and G. Lu, ChemSusChem, 2011, 4, 1758 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra22008c |
| This journal is © The Royal Society of Chemistry 2012 |