Selectivity control during the solid supported protic acids catalysed synthesis of 1,2-disubstituted benzimidazoles and mechanistic insight to rationalize selectivity†
Dinesh Kumar, Damodara N. Kommi, Rajesh Chebolu, Sanjeev K. Garg, Raj Kumar and Asit K. Chakraborti*
Department of Medicinal Chemistry, National Institute of Pharmaceutical Education and Research (NIPER), Sector 67, S. A. S. Nagar 160 062, Punjab, India. E-mail: akchakraborti@niper.ac.in
First published on 22nd October 2012
Abstract
Selectivity control during the formation of 1,2-disubstituted benzimidazoles has been achieved for the reaction of o-phenylenediamine with aldehydes in the presence of solid supported protic acids as catalysts and choosing an appropriate reaction medium. Perchloric acid adsorbed on silica-gel (HClO4–SiO2) was found to be the most effective catalyst system for the synthesis of 1,2-disubstituted benzimidazoles in EtOH at rt. Apart from the catalyst and solvent, the electronic and steric factors of the aldehyde and the electronic factor of the o-phenylenediamine are also significant contributory factors in dictating the selectivity. An understanding of the mechanistic course of the formation of the 1,2-disubstituted benzimidazoles has been outlined that would rationalise the origin of selectivity control under the set experimental parameters.
Introduction
In view of the role of the 1,2-disubstituted benzimidazole scaffold as a versatile pharmacophore for the generation of new chemical entities in diverse therapeutic areas,1 the formation of these privileged class of compounds has generated interest to develop newer synthetic methods.The various synthetic strategies (Scheme 1) adopted for the generation of this heterocyclic framework can be summarized by the following distinct routes: (i) cyclodehydration of N-alkyl-N-acyl-o-phenylenediamine (route A),1h,1i (ii) oxidative cyclocondensation of N-alkyl-o-phenylenediamine with aldehyde1h,2 (route B), (iii) N-alkylation of 2-substituted benzimidazole1b,d,g,k (route C), (iv) Suzuki coupling of aryl boronic acids with 1-iodo-2-alkyl benzimidazoles3 (route D), (v) copper-catalyzed amidation of o-halo N-alkylated anilines followed by cyclodehydration of the intermediately formed N-alkyl-N-acyl o-phenylenediamine4 (route E), (vi) copper/palladium-catalyzed amination of o-halo N-acylated anilines followed by cyclodehydration of the intermediately formed N-alkyl-N-acyl o-phenylenediamine5 (route F), (vii) palladium-catalysed intramolecular aryl-amination of (o-bromo/iodophenyl)amidines6 or bis(trifluoroacetoxy)iodobenzene mediated intramolecular cyclisation of N-alkyl-N′-arylamidines7 (route G). While the Buchwald and Suzuki chemistries (routes D–G) are used quite often by medicinal chemists, they require special efforts to prepare the desired starting materials.
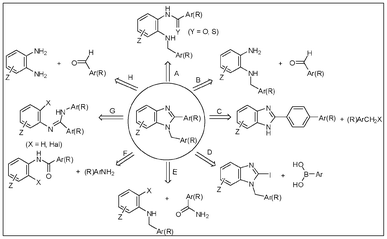 | ||
| Scheme 1 Synthetic strategies for the formation of 1,2-disubstituted benzimidazoles. | ||
Thus, a direct one-pot cyclocondensation of o-phenylenediamine with aldehydes (route H, Scheme 1) to prepare 1,2-disubstituted8 benzimidazoles appears to be a straightforward approach and has received unabated attention from organic/medicinal chemists for the generation of new hits/leads for potential therapeutic applications that require a convenient, selective and high yielding synthetic method to meet the needs of the timely supply of designed molecules for biological evaluation.9
However, the synthetic design following route H poses a potential selectivity problem due to the possibility of competitive formation of the 1,2-disubstituted and the 2-substituted benzimidazoles, an issue that remains inadequately addressed so far. The present work aims to control the selectivity in the formation of 1,2-disubstituted benzimidazoles during the cyclocondensation of o-phenylenediamines with aldehydes and offer a mechanistic understanding that would rationalise the selectivity control.
In view of the considerable awareness that has been generated for chemical processes on solid surfaces10 and the implication of heterogeneous catalysts (solid acids) in devising greener synthetic processes/methods,11 we describe herein a convenient and highly selective synthesis of 1,2-disubstituted benzimidazoles catalysed by solid acids such as HClO4–SiO2.12
Results and discussion
In a model study (Scheme 2), the cyclocondensation of o-phenylenediamine (1a) with two molar equivalents of 4-dimethylaminobenzaldehyde (2a) was performed separately in the presence of various catalyst systems. The use of HClO4–SiO2 (0.5 mol%) as the heterogeneous catalyst in EtOH at rt (25–30 °C) resulted in the formation of the desired 1,2-disubstituted benzimidazole 1-(4-N,N-dimethylaminophenylmethyl)-2-(4-N,N-dimethylphenyl)benzimidazole (3a) in 90% yield (Table 1).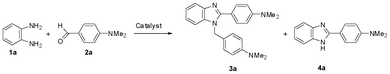 | ||
| Scheme 2 Selectivity of the formation of the 1,2-disubstituted benzimidazole 3a and the 2-substituted benzimidazole 4a during the reaction of 1a with 2a in the presence of various catalyst systems. | ||
| Entry | Catalyst (mol%) | Time (h) | Yield (%)b,c | Lit ref. | |
|---|---|---|---|---|---|
| 3a | 4a | ||||
| a 1a (2.5 mmol) was treated with 2a (5 mmol, 2 equiv.) in the presence of the catalyst (except for entries 1 and 2) in EtOH (5 mL) (except for entries 1, 5, 8, 11, 12, 13, 14, 15, 16 and 17.) at rt (∼25–30 °C).b The isolated yield of 3a and 4a obtained after column chromatographic purification.c The products were characterised by NMR (1H and 13C) and MS (APCI).d The reaction was carried out at 130 °C (oil bath) under neat conditions in the absence of any catalyst.e The products 3a and 4a were obtained in 27 and 20% yield, respectively, on performing the reaction for 4 h.f The reaction was carried out at rt in EtOH in the absence of any catalyst.g The products 3a and 4a were obtained in 30 and 20% yield, respectively, on performing the reaction for 4 h.h A 10% w/w of the catalyst was used.i The reaction was carried out under microwave heating (600 W output) in a domestic microwave oven under neat conditions.j The products 3a and 4a were obtained in 65 and 25% yield, respectively, on performing the reaction for 1.5 h.k The products 3a and 4a were obtained in 75 and 15% yield, respectively, on performing the reaction for 7 h.l The reaction was carried out in CHCl3 instead of EtOH.m The products 3a and 4a were obtained in 70 and 18% yield, respectively, on performing the reaction for 7.5 h following a reported9q procedure.n The reaction was carried out at rt under neat conditions.o The products 3a and 4a were obtained in 50 and 20% yield, respectively, on performing the reaction for 4 h at rt under neat conditions.p The reaction was carried out under neat conditions at 80 °C.q The reaction was carried out in water at 80 °C.r The products 3a and 4a were obtained in 65 and 20% yield, respectively, on performing the reaction for 3 h in water at 80 °C.s The reaction was carried out at rt.t The products 3a and 4a were obtained in 66 and 22% yield, respectively, on performing the reaction for 2 h at rt.u The products 3a and 4a were obtained in 73 and 10% yield, respectively, on performing the reaction for 5 h at rt.v The reaction was carried out in water at rt.w The products 3a and 4a were obtained in 72 and 16% yield, respectively, on performing the reaction in water at rt for 5 h. | |||||
| 1 | noned | 1 | trace | traces | this worke |
| 2 | nonef | 1 | trace | traces | this workg |
| 3 | HClO4–SiO2 (0.5) | 1 | 90 | 00 | this work |
| 4 | montmorillonite K-10 (10)h | 1 | 12 | 07 | this work using the reported catalyst8s |
| 5 | montmorillonite K-10 (10)h | 4 | 42 | 22 | this work using reported catalyst8s for the time reported therein |
| 6 | montmorillonite K-10 (10)h,i | 10 min | 80 | 10 | 8s |
| 7 | H2SO4–SiO2 (10) | 1 | 37 | 15 | this work using the reported catalystj8b |
| 8 | L-Proline (10) | 1 | 15 | traces | this work using the reported catalystk8q |
| 9 | L-Proline (10)l | 1 | 12 | traces | 8qm |
| 10 | oxalic acid (10) | 1 | 26 | traces | this work using the reported catalyst8d |
| 11 | oxalic acid (10) | 3 | 70 | 14 | this work using reported catalyst8d for the time reported therein |
| 12 | Fe(ClO4)3 (10) | 1 | 25 | traces | this work using the reported catalyst8e |
| 13 | Fe(ClO4)3 (10) | 3 | 72 | 10 | this work using reported catalyst8e for the time reported therein |
| 14 | Fe(ClO4)3 (10)n | 1 | 14 | 06 | 8eo |
| 15 | Mg(HSO4)2 (30)p | 20 min | 63 | 25 | 8p |
| 16 | Mg(HSO4)2 (30)q | 1 | 25 | 08 | 8pr |
| 17 | SiO2/ZnCl2 (25)n | 1 | trace | traces | 8n |
| 18 | amberlite-IR-20 (0.1 g)s | 1 | 35 | 10 | 8mt |
| 19 | Me3SiCl (50)s | 1 | 16 | traces | 8iu |
| 20 | [Hmim][TFA] (10)v | 1 | 15 | traces | 8hw |
The efficiency of HClO4–SiO2 was compared with the reported catalysts/processes. After the complete consumption of the starting materials, two new spots appeared in the TLC. The crude reaction mixtures were purified by column chromatography and the products eluted at different time intervals were identified as 3a and 2-(4-N,N-dimethylaminophenyl)benzimidazole (4a) (Scheme 2 and Table 1). However, when HClO4–SiO2 was used as the catalyst, the crude product mixtures exhibited a single spot in the TLC and 3a was obtained as the only product after purification either by crystallisation or column chromatography. The superiority of HClO4–SiO2 was clearly established both in terms of the product yields as well as the selectivity by comparing the results obtained for the reaction of 1a with 2a with those obtained using reported catalysts. While the use of HClO4–SiO2 afforded 3a as the sole product in 90% yield, the reported catalysts were found to be less effective and lacked selectivity affording 3a in 42–80% yields along with the formation of 4a in 10–25% yields. The use of a catalyst was found to be essential as inferior yields and selectivity were observed on performing the reaction in the absence of any catalyst (entries 1 and 2, Table 1).
The reaction medium appears to have a significant influence in controlling the selective formation of the 1,2-disubstituted benzimidazole as evidenced by the results (product yield and selectivity) during the HClO4–SiO2 catalysed cyclocondensation of 1a with 2a performed in various protic polar, aprotic polar, halogenated, weakly polar (ethereal), and non-polar (hydrocarbon) solvents as well as under neat conditions.13 The use of non polar, weakly polar, aprotic polar solvents and neat conditions decreased the selectivity and in each case 3a was associated with the formation of 4a. The best results were obtained in EtOH but the selectivity decreased when using other protic solvents such as MeOH, iPrOH, tBuOH, ethylene glycol and water. Inferior results were also obtained with PEG-200, DCE and PhMe.
The efficiency of HClO4–SiO2, in comparison to that of other protic acids adsorbed on silica gel, is demonstrated by the results incorporated in Table 2. However, for this study for the reaction of 1a we took 4-methylbenzaldeheyde (2b) instead of 2a so as to avoid any influence of salt formation with the NMe2 group of 2a on the selectivity and product yield (Scheme 3).
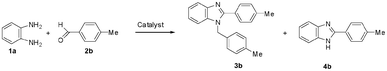 | ||
| Scheme 3 Selectivity of the formation of the 1,2-disubstituted benzimidazole 3b and the 2-substituted benzimidazole 4b during the reaction of 1a with 2b in the presence of various catalysts. | ||
| Entry | Catalyst (mol%) | Yield (%)b,c | |
|---|---|---|---|
| 3b | 4b | ||
| a 1a (2.5 mmol) was treated with 2b (5 mmol, 2 equiv.) in the presence of the catalyst in EtOH (5 mL except for entries 8–10) at rt (∼25–30 °C) for 1.5 h unless specified.b The isolated yield of 3b and 4b after column chromatographic purification.c The products were characterised by NMR (1H and 13C) and MS (APCI).d The reaction was carried out in water.e The reaction was carried out in 1 M (5 mL) aq LiCl.f The reaction was carried out in 1 M (5 mL) aq LiCl in the absence of any added catalyst.g A 10% w/w of the catalyst was used. | |||
| 1 | HClO4–SiO2 [230–400] (0.5) | 80 | 5 |
| 2 | HClO4–SiO2 [100–200] (0.5) | 55 | 09 |
| 3 | HClO4–SiO2 [60–120] (0.5) | 18 | 10 |
| 4 | TfOH–SiO2 (1) | 61 | 12 |
| 5 | TFA–SiO2 (1) | 48 | 13 |
| 6 | p-TsOH–SiO2 (1) | 58 | 20 |
| 7 | MSA–SiO2 (1) | 55 | 12 |
| 8 | HBr–SiO2 (1) | 54 | 15 |
| 9 | H2SO4–SiO2 (10) | 40 | 08 |
| 10 | H2SO4–SiO2 (10)d | 42 | 10e |
| 11 | H2SO4–SiO2 (10)e | 38 | 08 |
| 12 | —f | trace | trace |
| 13 | HClO4–neutral Al2O3 (1) | 45 | 12 |
| 14 | HClO4–basic Al2O3 (1) | 48 | 15 |
| 15 | HClO4–acidic Al2O3 (1) | 55 | 12 |
| 16 | zeolite type Y (10)g | 25 | 10 |
| 17 | zeolite K L−1 (SAR 6.8)(10)g | 21 | 10 |
| 18 | zeolite ZSM 5 (SAR 6.8) (10)g | 28 | 08 |
| 19 | zeolite K L−1 (for synthesis) (10)g | 21 | 08 |
| 20 | zeolite Na/Fau(10)g | 20 | 08 |
| 21 | zeolite NH4/Y(10)g | 24 | 13 |
| 22 | montmorillonite K-10 (10)g | 15 | 09 |
| 23 | montmorillonite KSF (10)g | 12 | 08 |
| 24 | SiO2 [230–400] (10)g | trace | trace |
| 25 | SiO2 [100–200] (10)g | trace | trace |
| 26 | SiO2 [60–120] (10)g | trace | trace |
| 27 | amberlite (10)g | trace | trace |
The nature of the solid support was also found to be important in maintaining the selectivity as the use of neutral, basic or acidic alumina instead of silica-gel as the solid support decreased the selectivity inducing competitive formation of 4b in 18–30% yields (entries 16–21, Table 2). Amongst the different varieties of silica gel used as the support, the 230–400 mesh size (henceforth in the text SiO2 refers to the 230–400 mesh size silica gel only) proved to be the most effective support (entries 1–3, Table 2). As silica and montmorillonite clays themselves have the ability to catalyse various organic reactions14 the catalytic efficiency of the solid support (various forms of silica gel) itself (without taking any protic acid) and other heterogeneous catalysts such as zeolites, clays, and amberlite (entries 16–27, Table 2) were tested and found to be inferior due to product yields and poor selectivity.
The efficiency of other inorganic and organic protic acids adsorbed on SiO2 was compared with that of HClO4–SiO2 for the reaction of 1a with 4-methylbenzaldeheyde (2b) (Scheme 3). Although good to excellent cyclocondensation took place with organic (entries 4–7, Table 2), other inorganic protic acids (entries 8–11, Table 2), and aq LiCl (in the presence/absence of any protic acid), the selectivity was inferior as the 2-substituted product 4b was formed in 15–35% yields in addition to the 1,2-disubstituted product 3b (15–72%). The best performance in terms of the amount of the catalyst, reaction time, product yield and 3b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4b selectivity was exhibited by HClO4–SiO2.13 The distinct advantage of using the supported protic acids instead of the protic acids as such was revealed during the reaction of 1a with 2b performed under the catalytic influence of the individual protic acids. In each case, the overall yield of the cyclocondensation products as well as the 3b:4b selectivity was found to be inferior to the corresponding reactions performed using the protic acids supported on SiO2.
:4b selectivity was exhibited by HClO4–SiO2.13 The distinct advantage of using the supported protic acids instead of the protic acids as such was revealed during the reaction of 1a with 2b performed under the catalytic influence of the individual protic acids. In each case, the overall yield of the cyclocondensation products as well as the 3b:4b selectivity was found to be inferior to the corresponding reactions performed using the protic acids supported on SiO2.
The generality of the HClO4–SiO2 catalysed selective formation of 1,2-disubstituted benzimidazole is demonstrated through the reaction of 1a with various aromatic, heteroaromatic, alicyclic and aliphatic aldehydes (Table 3). The desired 1,2-disubstituted benzimidazoles were obtained in 70–90% yields. However, the steric and electronic nature of the substrate (aldehyde) exhibited a significant influence on the outcome of the selectivity. Sterically hindered (entries 9 and 20, Table 3) and electron deficient (entry 10, Table 3) aldehydes afforded exclusively the 2-substituted benzimidazole. The catalysts were easily separated from the products by filtration. The isolated products were purified by crystallisation from aq EtOH or by passing through a column of silica-gel and eluting with 10% EtOAc in heptane, wherever required.
| Entry | Substrate | Time (h) | Yield (%)b,c |
|---|---|---|---|
| a 1a (2.5 mmol) was treated with the aldehyde (5 mmol, 2 equiv.) in EtOH (5 mL) separately in the presence of HClO4–SiO2 (0.5 mol%) at rt (∼25–30 °C).b The isolated yield of the corresponding 1,2-disubstituted benzimidazole after column chromatographic purification.c The products were characterised by NMR (1H and 13C) and MS (APCI).d The corresponding 2-substituted benzimidazole was the sole product. | |||
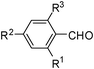 | |||
| 1 | R1 = R2 = R3 = H | 2 | 80 |
| 2 | R1 = R3 = H; R2 = Me | 1.5 | 80 |
| 3 | R1 = R3 = H; R2 = OMe | 1 | 90 |
| 4 | R1 = R3 = H; R2 = NMe2 | 1 | 90 |
| 5 | R1 = R3 = H; R2 = Cl | 2 | 85 |
| 6 | R1 = R3 = H; R2 = Br | 2 | 86 |
| 7 | R1 = R3 = H; R2 = CF3 | 2 | 85 |
| 8 | R1 = R3 = H; R2 = OCH2Ph | 2 | 80 |
| 9 | R1 = R2 = R3 = Me | 3 | 75d |
| 10 | R1 = R3 = H; R2 = NO2 | 2.5 | 86d |
| 11 | 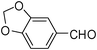 | 1 | 90 |
| 12 | 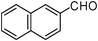 | 2 | 90 |
| 13 | 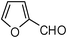 | 2 | 75 |
| 14 | 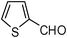 | 1 | 80 |
| 15 | 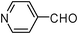 | 2 | 75 |
| 16 | 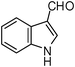 | 3 | 75 |
| 17 | 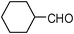 | 2 | 80 |
| 18 | 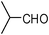 | 2 | 75 |
| 19 | 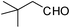 | 2 | 78 |
| 20 | 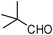 | 3 | 75d |
The recyclability of the catalyst was investigated during the reaction of 1a with 2b.13 After the completion of the reaction, the catalysts were recovered by filtration and reused for a subsequent fresh batch of the reaction after reactivation. The catalytic activity was found to be retained up to a fifth run in carrying out the reactions in 50, 40, 30, 20, and 10 mmol scale with respect to 1a affording 90, 90, 87, 85, and 85% yields with HClO4–SiO2. The feasibility for the large scale preparation of 1,2-disubstituted benzimidazole was demonstrated with 100 mmol scale reactions.
To rationalise the influence of the catalyst, solvent, and the steric and electronic nature of the substrate on the selectivity control, we set forth to understand the mechanistic course of the reaction. The cyclocondensation of o-phenylenediamines with aldehydes to form the 1,2-disubstituted benzimidazoles may proceed through two distinctly different pathways (Scheme 4): (i) a bis-imine-rearrangement route (path a)8a,d,e,i,k,n,o,u and (ii) a mono-imine-cyclocondensation-aminal/immonium-rearrangement route (path b).8l,m,p ‘Path a’, would involve the formation of the bis-imine IIa (this may arise in a stepwise fashion of mono-imine Ia formation followed by the formation of the bis-imine IIa), which may undergo rearrangement via intramolecular nucleophilic attack of the nitrogen electron lone pair of one of the imine groups to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N of the other imine group to from IIIa followed by a 1,3-hydride shift to generate the 1,2-disubstituted benzimidazole 3. In the alternative route ‘path b,’ the reaction may proceed through the formation of the mono-imine Ia, followed by intramolecular nucleophilic attack of the adjacent amino group on the imine to form the benzimidazolidine Ib, which would react with another molecule of the aldehyde to form the aminal/immonium species IIb/IIIb that on 1,3-hydrogen shift would lead to 3.
N of the other imine group to from IIIa followed by a 1,3-hydride shift to generate the 1,2-disubstituted benzimidazole 3. In the alternative route ‘path b,’ the reaction may proceed through the formation of the mono-imine Ia, followed by intramolecular nucleophilic attack of the adjacent amino group on the imine to form the benzimidazolidine Ib, which would react with another molecule of the aldehyde to form the aminal/immonium species IIb/IIIb that on 1,3-hydrogen shift would lead to 3.
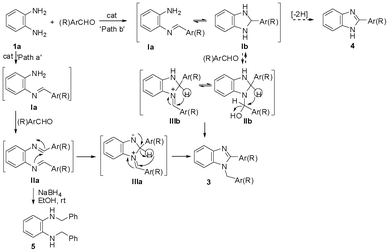 | ||
| Scheme 4 Pathways of 1,2-disubstituted benzimidazole formation during the HClO4–SiO2 catalysed reaction of o-phenylenediamine with aldehyde. | ||
The involvement of the 1,3-hydrogen shift was corroborated by the formation of 2-phenyl-1-α-d2-phenylmethyl-1H-benzimidazole 3c–d8i during the reaction of deuterated benzaldehyde 2c–d (2 molar equiv.) with 1a (Scheme 5). However, the feasibility of the formation of 3 by ‘path a’ or ‘path b’ could not be distinguished.
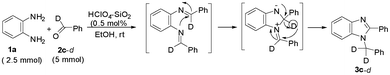 | ||
| Scheme 5 Evidence for the 1,3-hydrogen shift during the formation of 2-phenyl-1-α-d2-methylphenyl-1H-benzimidazole by the HClO4–SiO2-catalysed reaction of o-phenylenediamine with deuterated benzaldehyde. | ||
Thus, to demonstrate the mechanistic route for the progress of the formation of the 1,2-disubstituted benzimidazole, we planned to intercept the possible intermediates by various spectrometric studies. The formation of the 1,2-disubstituted benzimidazole via ‘path b’ seems unlikely as no ion peak corresponding to either of the aminal IIb or the immonium species IIIb (expected to be formed through the condensation of Ib with another molecule of the aldehyde) was detected (GCMS and LCMS) during the progress of the reaction of 1a with 2b (2 equiv.). The intermediate formation of Ib would further imply that the dehydrogenative conversion to the undesired product 2-substituted benzimidazole 4 would be accelerated under the presence of air/oxygen and on the other hand would be retarded under anaerobic conditions.8l The treatment of 1a with 2b (2 equiv.) under the catalytic influence of HClO4–SiO2 afforded 3b in 78% yield using deoxygenated EtOH under nitrogen atmosphere compared to an 80% yield obtained in using EtOH in open air. Further, 3b was formed in 80% yield during the HClO4–SiO2 catalysed reaction of 1a with 2b (2 equiv.) in EtOH while oxygen gas was bubbled into the reaction mixture. Thus, the presence or the absence of air/oxygen did not have any significant influence on the overall outcome of the reaction. These ruled out the involvement of Ib (‘path b’) during 1,2-disubstituted benzimidazole formation for which the reaction follows ‘path a’.
To further demonstrate the progress of the reaction for 1,2-disubstituted benzimidazole formation via ‘path a,’ we tested the feasibility of imine formation under the adopted experimental procedure. Treatment of aniline with benzaldehyde 2c under the catalytic influence of HClO4–SiO2 under similar conditions formed the imine. Bis-imine formation also takes place when m-phenylenediamine is treated with two molar equivalents of 2c in the presence of HClO4–SiO2 under similar conditions. These suggested that the reaction might proceed via ‘path a’ involving the formation of the bis-imine IIa. As a direct proof, the progress of the reaction of 1a with 2c (2 equiv.) was monitored by GCMS at 10, 20, 30, 40, and 50 min time intervals to ‘fish out’ any relevant intermediate. In each case, the ion peak at m/z 312 was detected. However, it remained unclear whether this could be due to the bis-imine IIa (Ar = Ph) or the corresponding 1,2-disubstituted benzimidazole (1-phenylmethyl-2-phenyl-1H-benzimidazole 3c) as both are of the same molecular weight. Detailed MSn studies also proved to be inconsequential with respect to the identity/authenticity of the species of m/z 312 as IIa (Ar = Ph) or 3c. However, when an aliquot of sample withdrawn from the reaction mixture of 1a with 2c (2 equiv.) was subjected to LCMS studies during a 5–60 min reaction period, the ion peak corresponding to the mono-imine of 1a and 2c could be detected. The height/area of this ion peak was found to decrease with time with a concomitant appearance of the peak at m/z 312 corresponding to the bis-imine with increasing intensity.
Finally, the formation of 1,2-disubstituted benzimidazole via ‘path a’ was validated by the isolation and characterisation (NMR) of N1,N2-dibenzylbenzene-1,2-diamine (5) formed by the in situ reduction (NaBH4) of the intermediately formed bis-imine (Scheme 4) during the course of the reaction between 1a and 2c providing for the first time crucial evidence that the reaction proceeds via path a.
Therefore, the selectivity of the 1,2-disubstituted and 2-substituted benzimidazole formation lies in the feasibility of the formation of the bis-imine under the prescribed set of experimental condition(s). A stronger catalyst (supported protic acid) and polar reaction medium should assist in bis-imine formation. Hence, EtOH (protic polar) is the most effective reaction medium for the 1,2-disubstituted benzimidazole formation. The intermediacy of the bis-imine IIa is the crucial and determining factor for 1,2-disubstituted benzimidazole formation and hence directing/controlling the 1,2-disubstituted vs. 2-substituted benzimidazole selectivity is demonstrated by the observation that the reaction of 1a with two molar equivalents of 2,4,6-trimethylbenzaldehyde 2d forms only the 2-(2,4,6-trimethyl)phenyl-1H-benzimidazole 4d in EtOH in the presence of HClO4–SiO2 (entry 9, Table 3). Due to the steric crowding surrounding the aldehyde group in 2d, the bis-imine is not feasible and the 2-substituted benzimidazole 4d is formed exclusively. To demonstrate that the reaction of 1a with 2d only forms the mono-imine (which on intramolecular nucleophilic attack by the adjacent amino group results in the formation of 4dvia the corresponding imidazoline), the reaction mixture was subjected to treatment with NaBH4 (Scheme 6). The formation of 2-N-(2,4,6-trimethyl)aminomethylaniline 6 justified that the lack of formation of the expected 1,2-disubstituted benzimidazole from the reaction of 1a with 2d is due to the feasibility of only mono-imine formation. The implication of the steric factor in suppressing the bis-imine formation (and hence directing the selectivity towards the formation of 2-substituted benzimidazole) can also be realized with a sterically hindered aliphatic aldehyde. Thus, the reaction of 1a with pivalaldehyde (2 equiv.) under the catalytic influence of HClO4–SiO2 in EtOH forms exclusively the 2-substituted benzimidazole (entry 20, Table 3), as the steric effect of the tBu group in pivalaldehyde does not permit the bis-imine formation with 1a.
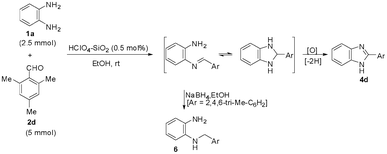 | ||
| Scheme 6 Formation of 2-substituted benzimidazole during the HClO4–SiO2 catalysed reaction of 1a with 2d. | ||
The selective formation of the 2-substituted benzimidazole from an electron deficient aldehyde such as 4-nitrobenzaldehyde 2e (entry 10, Table 3), under conditions conducive to form 1,2-disubstituted benzimidazole, could be due to the fact that because of the highly electrophilic character of the aldehyde carbonyl, rapid formation of the mono-imine takes place and before the second amino group of 1a is involved in imine formation with another molecule of 2e it undergoes an intramolecular nucleophilic attack on the CN of the mono-imine (due its high electrophilic character owing to the 4-nitro group) leading to the formation of the intermediate imidazoline and subsequent dehydrogenation to the 2-substituted benzimidazole as the final/end product.
To assess the influence of the electronic effect of the o-phenylenediamine component on the selectivity of formation of the 1,2-disubstituted and 2-substituted benzimidazoles, 4-nitro-o-phenylenediamine 1b was treated separately with 2 equiv. each of benzaldehyde 2c, 4-nitrobenzaldehyde 2e, and 4-methoxybenzaldehyde 2f (Scheme 7) under conditions that would favour the formation of 1,2-disubstituted benzimidazole. However, in each case the corresponding 2-substituted benzimidazoles were obtained as the only product.
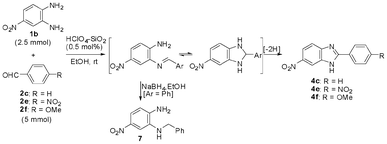 | ||
| Scheme 7 HClO4–SiO2-catalysed reaction of 4-nitro-o-phenylenediamine with 2c, 2e, and 2f. | ||
The origin of the selective formation of the 2-substituted benzimidazoles from the reactions of 1b could be due to its ability to form the mono-imine only as the electronic withdrawing effect of the nitro group makes the para amino group less nucleophilic. To prove that the reaction of 1b with an aldehyde can only form the mono-imine, the reaction mixture of 1b and 2c was treated with NaBH4 to trap any intermediately formed imine by converting it to the corresponding reduced product which was found to be 4-nitro-2-methylphenylaminoaniline 7. The formation of the mono-N-benzylated product of 1b confirmed that only the mono-imine is formed although 2 molar equiv. of the aldehyde was used. That the 2-amino group (meta to the nitro group) in 1b undergoes imine formation was confirmed by comparison of the 1H NMR (the benzylic protons being the probe nuclei) of the reductive alkylation product from the reaction of 1b and 2c with that of the N-benzylated 4- and 3-nitro anilines (prepared by reductive amination of 4- and 3- nitro anilines with benzaldehyde). The benzylic protons in N-benzyl-4-nitroaniline and N-benzyl-3-nitroaniline appear at δ 4.43 and 4.39, respectively. The benzyl protons of the reductive alkylation product obtained from the reaction of 1b with 2c appeared at δ 4.38 suggesting it to be 7 (Scheme 7). However, we realised that the tiny chemical shift difference between N-benzyl 4-nitro aniline and N-benzyl 3-nitro aniline cannot differentiate the regioisomers unambiguously. A recent literature report15 on the reductive amination of 1b and 2c came to our notice which, although, claimed the formation of 7, the ambiguity remains with respect to the identity of the product either as 7 or as 7a. In order to resolve this issue, we carried out various NMR experiments such as DEPT 135 and 2D (HSQC and NOESY) of 7 and confirmed its structure as 4-nitro-2-methylphenylaminoaniline (supporting information†).
Conclusions
This study provides an account on the issue of competitive formation of the 1,2-disubstituted and 2-substituted benzimidazoles during direct cyclocondensation of o-phenylenediamine aldehyde. The scope and limitation of various protic acids on a solid support as well as the reaction medium have been studied to derive that the best operative reaction conditions are the treatment of the o-phenylenediamine with two molar equiv. of an aldehyde in EtOH at rt in the presence of catalytic quantities of HClO4–SiO2 (0.5 mol%) to form exclusively/selectively the 1,2-disubstituted benzimidazoles. Other solid supports such as alumina (acidic/basic/neutral) gave inferior results both in terms of product yield and selectivity. The catalyst system HClO4–SiO2 is distinctly superior to the reported catalysts/procedures that are associated with the competitive formation of the 2-substituted benzimidazoles. A mechanistic outline has been derived for the HClO4–SiO2 catalysed formation of the 1,2-disubstituted benzimidazoles, which follows bis-imine formation, rearrangement, and a 1,3-hydride shift pathway. The origin/rationale of selectivity resides on furthering or suppressing the bis-imine formation using a judicial choice of the supported protic acid catalyst as well as an appropriate reaction medium. The steric and electronic factors of the aldehyde and the electronic factor of the o-phenylenediamine component also exhibit significant influence on the selectivity. Sterically hindered and electron deficient aldehydes and electron deficient o-phenylenediamine afforded the 2-substituted benzimidazoles under conditions that are conducive for 1,2-disubstituted benzimidazole formation.Preparation of perchloric acid adsorbed on silica-gel (HClO4–SiO2)
The HClO4–SiO2 was prepared following the procedure first reported by its inventors.12 To a suspension of silica gel (23.75 g, 230–400 mesh) in Et2O (50 mL), was added HClO4 (1.25 g, 12.5 mmol, 1.78 mL of a 70% aq solution of HClO4) and the mixture was stirred magnetically for 30 min at rt. The Et2O was removed under reduced pressure (rotary evaporator) and the residue heated at 100 °C for 72 h under vacuum (10 mm Hg) to afford HClO4–SiO2 (0.5 mmol g−1) as a free flowing powder.Typical experimental procedure for the synthesis of 2-aryl-1-arylmethyl-1H-1,3-benzimidazoles using HClO4–SiO2 (Entry 4, Table 3)
The mixture of o-phenylenediamine 1a (0.27 g, 2.5 mmol, 1 equiv.), 4-dimethylaminobenzaldehyde 2a (0.74 g, 5 mmol, 2 equiv.), and HClO4–SiO2 (25 mg, 0.012 mmol, 0.5 mol%) in EtOH (10 mL) was stirred magnetically at rt (∼25–30 °C). After completion of the reaction (TLC, 3:1 n-hexane–EtOAc), the mixture was filtered, washed with EtOH (2 × 5 mL), and the combined filtrates were concentrated under rotary vacuum evaporation. The crude product was purified by crystallisation from aq EtOH or passed through a column of silica gel and eluted with 10% EtOAc in hexane to afford 1-(4-dimethylaminophenylmethyl)-2-(4-dimethylaminophenyl)-1H-benzimidazole 3a (0.83 g, 90%) as a white solid; mp = 255–256 °C; IR (KBr) νmax = 3038, 1610, 1562, 1482, 1251, 1168 cm−1; 1H NMR (300 MHz, 25 °C, CDCl3): δ = 7.80 (d, J = 7.8 Hz, 2 H; Ar–H), 7.63 (d, J = 8.7 Hz, 2 H; Ar–H), 7.19 (t, J = 8.5 Hz, 3 H; Ar–H), 7.01 (d, J = 8.5 Hz, 2 H; Ar–H), 6.66–6.78 (m, 4 H; Ar–H), 5.37 (s, 2 H; CH2), 3.00 (s, 3 H; CH3), 2.92 (s, 3 H; CH3); MS (APCI) m/z: 371 (M+H)+.8cTypical experimental procedures for mechanistic insights
:15) to afford N1, N2-dibenzylbenzene-1,2-diamine 5 (86 mg, 12%). Low melting solid; IR (KBr) νmax = 3435, 2972, 1612, 1451, 1325, 1260 cm−1; 1H NMR (CDCl3, 400 MHz, TMS) δ: 7.39–7.36 (m, 4H), 7.34–7.29 (m, 4H), 7.27–7.24 (m, 2H), 6.80–6.77 (m, 2H), 6.73–6.70 (m, 2H), 4.31 (s, 4H), 3.64 (brs, 2H); MS (EI) m/z: 289.4 (M+H)+.:1 n-heptane–EtOAc), the mixture was filtered, washed with EtOH (2 × 5 mL), and the combined filtrates were concentrated under rotary vacuum evaporation. The crude product was purified by passing through a column of silica gel and eluting with 10% EtOAc in heptane to afford 2-phenyl-1-α-d2-phenylmethyl-1H-benzimidazole 3c–d (0.20 g, 72%) as a white solid; 1H NMR (400 MHz, 25 °C, CDCl3): δ = 7.87 (d, J = 2 Hz, 1 H; Ar–H), 7.70–7.68 (m, 2 H; Ar–H), 7.48–7.42 (m, 3 H; Ar–H), 7.36–7.29 (m, 4 H; Ar–H), 7.24–7.20 (m, 2 H; Ar–H), 7.12–7.10 (m, 2 H; Ar–H); 13C NMR (100 MHz, CDCl3): δ = 154.1, 143.2, 136.2, 136.0, 130.0, 129.9, 129.2, 129.0, 128.7, 127.8, 126.0, 123.0, 122.6, 120.0, 110.5; MS (APCI) m/z: 287.4 (M+H)+.8iNote added after first publication
This article replaces the version published on 6th November 2012, which contained errors in the title.Acknowledgements
DK and DNK thank CSIR, New Delhi for RA and SRF respectively. RC and SKG thank UGC for SRF.References
- (a) Angiotensin II receptor antagonist: Y. Kohara, K. Kubo, E. Imamiya, T. Wada, Y. Inada and T. Naka, J. Med. Chem., 1996, 39, 5228 CrossRef CAS; (b) Antiviral: A. R. Porcari, R. V. Devivar, L. S. Kucera, J. C. Drach and L. B. Townsend, J. Med. Chem., 1998, 41, 1252 CrossRef CAS; (c) Neuropeptide Y Y1 receptor antagonist: H. Zarrinmayeh, A. M. Nunes, P. L. Ornstein, D. M. Zimmerman, M. B. Arnold, D. A. Schober, S. L. Gackenheimer, R. F. Bruns, P. A. Hipskind, T. C. Britton, B. E. Cantrell and D. R. Gehlert, J. Med. Chem., 1998, 41, 2709 CrossRef CAS; (d) Factor Xa inhibitor: Z. S. Zhao, D. O. Arnaiz, B. Griedel, S. Sakata, J. L. Dallas, M. Whitlow, L. Trinh, J. Post, A. Liang, M. M. Morrissey and K. J. Shaw, Bioorg. Med. Chem. Lett., 2000, 10, 963 CrossRef CAS; (e) Antiallergic 5-Lipoxygenase inhibitor: H. Nakano, T. Inoue, N. Kawasaki, H. Miyakata, H. Matsumoto, T. Taguch, N. Inagaki, H. Nagai and T. Satog, Bioorg. Med. Chem., 2000, 8, 373 CrossRef CAS; (f) Antibacterial: H. Göker, C. Kuş, D. W. Boykin, S. Yildiz and N. Altanlar, Bioorg. Med. Chem., 2002, 10, 2589 CrossRef; (g) Antimicrobial: G. Ayhan-Kılcıgil and N. Altanlar, Farmaco, 2003, 58, 1345 CrossRef; (h) S. Özden, D. Atabey, S. Yildiz and H. Göker, Bioorg. Med. Chem., 2005, 13, 1587 CrossRef; (i) Anti-Hepatitis C virus agents: T. Ishida, T. Suzuki, S. Hirashima, K. Mizutani, A. Yoshida, I. Ando, S. Ikeda, T. Adachi and H. Hashimoto, Bioorg. Med. Chem. Lett., 2006, 16, 1859 CrossRef CAS; (j) J. R. Hwu, R. Singha, S. C. Hong, Y. H. Chang, A. R. Das, I. Vliegen, E. De Clercq and J. Neyts, Antiviral Res., 2008, 77, 157 CrossRef CAS; (k) Burkitt's lymphoma promotion inhibitors: M. M. Ramla, M. A. Omar, H. Tokuda and H. I. El-Diwani, Bioorg. Med. Chem., 2007, 15, 6489 CrossRef CAS; (l) Antimicrobial and antiviral agents: D. Sharma, B. Narasimhan, P. Kumar, V. Judge, R. Narang, E. De Clercq and J. J. Balzarini, J. Enzyme Inhib. Med. Chem., 2009, 24, 1161 CrossRef CAS; (m) Antiparasitic agents: J. Pérez-Villanueva, R. Santos, A. Hernández-Campos, M. A. Giulianotti, R. Castillo and J. L. Medina-Franco, Med. Chem. Commun., 2011, 2, 44 RSC.
- (a) A. Mazurov, Bioorg. Med. Chem. Lett., 2000, 10, 67 CrossRef CAS; (b) D. Tumelty, M. K. Schwarz and M. C. Needels, Tetrahedron Lett., 1998, 39, 7467 CrossRef CAS and the references therein.
- (a) J. M. Smith and V. Krchňák, Tetrahedron Lett., 1999, 40, 7633 CrossRef CAS; (b) D. Vourloumis, M. Takahashi, K. B. Simonsen, B. K. Ayida, S. Barluenga, G. C. Winters and T. Hermann, Tetrahedron Lett., 2003, 44, 2807 CrossRef CAS.
- J. Ezquerra and C. Lamas, Tetrahedron, 1997, 53, 12755 CrossRef CAS.
- N. Zheng and S. L. Buchwald, Org. Lett., 2007, 9, 4749 CrossRef CAS.
- (a) B. Zou, Q. Yuan and D. Ma, Angew. Chem., Int. Ed., 2007, 46, 2598 CrossRef CAS; (b) N. Zheng, K. W. Anderson, X. Huang, N. H. Nguyen and S. L. Buchwald, Angew. Chem., Int. Ed., 2007, 46, 7509 CrossRef CAS.
- (a) C. T. Brain and S. A. Brunton, Tetrahedron Lett., 2002, 43, 1893 CrossRef CAS; (b) C. T. Brain and J. T. Steer, J. Org. Chem., 2003, 68, 6814 CrossRef CAS; (c) P. Saha, M. A. Ali, P. Ghosh and T. Punniyamurthy, Org. Biomol. Chem., 2010, 8, 5692 RSC.
- (a) A. Rao, A. Chimirri, S. Ferro, A. M. Monforte, P. Monforte and M. Zappalà, ARKIVOC, 2004, V, 147 Search PubMed; (b) P. Salehi, M. Dabiri, M. A. Zolfigol, S. Otokesh and M. T. Baghbanzadeh, Tetrahedron Lett., 2006, 47, 2557 CrossRef CAS; (c) R. Varala, A. Nasreen, R. Enugala and S. R. T. Adapa, Tetrahedron Lett., 2007, 48, 69 CrossRef CAS; (d) N. D. Kokare, J. N. Sangshetti and D. B. Shinde, Synthesis, 2007, 2829 CAS; (e) H. A. Oskooie, M. M. Heravi, A. Sadnia, F. K. Behbahani and F. Janati, Chin. Chem. Lett., 2007, 18, 1357 CrossRef CAS; (f) S. S. Pawar, D. V. Dekhane, M. S. Shingare and S. N. Thore, Chin. Chem. Lett., 2008, 19, 1055 CrossRef CAS; (g) M. Dabiri, P. Salehi, M. Baghbanzadeh and M. S. Nikcheh, Synth. Commun., 2008, 38, 4272 CrossRef CAS; (h) K. Niknam, M. A. Zolfigol and N. Safikhani, Synth. Commun., 2008, 38, 2919 CrossRef CAS; (i) J. -P. Wan, S. -F. Gan, J. -M. Wu and Y. Pan, Green Chem., 2009, 11, 1633 RSC; (j) A. A. Mohammadi, J. Azizian and N. Karimi, Heterocycles, 2009, 78, 2337 CrossRef CAS; (k) G. R. Jadhav, M. U. Shaikh, R. P. Kale and C. H. Gill, Chin. Chem. Lett., 2009, 20, 535 CrossRef CAS; (l) D. Saha, A. Saha and B. C. Ranu, Green Chem., 2009, 11, 733 RSC; (m) S. Das Sharma and D. Konwar, Synth. Commun., 2009, 39, 980 CrossRef CAS; (n) R. G. Jacob, L. G. Dutra, C. S. Radatz, S. R. Mendes, G. Perin and E. J. Lenardao, Tetrahedron Lett., 2009, 50, 1495 CrossRef CAS; (o) K. Bahrami, M. K. Mohamma and A. Nejati, Green Chem., 2010, 12, 1237 RSC; (p) N. Khodabakhsh, M. A. Zolfigol and N. Safikhani, Synth. Commun., 2008, 38, 2919 CrossRef; (q) J. S. Yadav, B. V. S. Reddy, K. Premalatha and S. K. Shiva, Can. J. Chem., 2008, 86, 124 CrossRef CAS; (r) S. Perumal, S. Mariappan and S. Selvaraj, ARKIVOC, 2004, VIII, 46 Search PubMed; (s) G. R. Jacob, G. L. Dutra, S. C. Radatz, R. S. Mendes, G. Perin and J. E. Lenardao, Tetrahedron Lett., 2009, 50, 1495 CrossRef; (t) M. Chakrabarty, R. Mukherjee, S. Karmakar and Y. Harigaya, Monatsh. Chem., 2007, 138, 1279 CrossRef CAS; (u) L.-J. Zhang, J. Xia, Y.-Q. Zhou, H. Wang and S.-W. Wang, Synth. Commun., 2012, 42, 328 CrossRef CAS; (v) S. Paul and B. Basu, Tetrahedron Lett., 2012, 53, 4130 CrossRef CAS.
- J. Potosky, Drug Discovery Today, 2005, 10, 115 CrossRef.
- (a) G. Ertl, Angew. Chem., Int. Ed., 2008, 47, 3524 CrossRef CAS; (b) The 2007 Nobel Prize in Chemistry.
- (a) K. Wilson and J. H. Clark, Pure Appl. Chem., 2000, 72, 1313 CrossRef CAS; (b) A. Corma and H. García, Chem. Rev., 2003, 103, 4307 CrossRef CAS; (c) J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37 CrossRef.
- (a) The heterogeneous catalyst systems HClO4–SiO2 and HBF4–SiO2 were/are not commercially available and were prepared and used for the first time by Chakraborti et al.: A. K. Chakraborti and R. Gulhane, Chem. Commun., 2003, 1896 RSC; (b) A. K. Chakraborti and R. Gulhane, Tetrahedron Lett., 2003, 44, 3521 CrossRef CAS; (c) A. K. Chakraborti and R. Gulhane, Indian Patent, 248506; Date: 20-07-2011. Appl. No. 266/DEL/2003. Filing Date: 10-3-2003 Search PubMed.
- See supporting information.
- (a) G. Sharma, R. Kumar and A. K. Chakraborti, J. Mol. Catal. A: Chem., 2007, 263, 143 CrossRef CAS; (b) S. V. Chankeshwara and A. K. Chakraborti, J. Mol. Catal. A: Chem., 2006, 253, 198 CrossRef CAS; (c) A. K. Chakraborti, A. Kondaskar and S. Rudrawar, Tetrahedron, 2004, 60, 9085 CrossRef CAS; (d) A. K. Chakraborti, S. Rudrawar and A. Kondaskar, Org. Biomol. Chem., 2004, 2, 1277 RSC.
- S. Dalapati, S. Jana, R. Saha, M. A. Alam and N. Guchhait, Org. Lett., 2012, 14, 3244 CrossRef CAS.
- (a) A. F. Abdel-Magid, K. G. Carson, B. D. Harris, C. A. Maryanoff and R. D. Shah, J. Org. Chem., 1996, 61, 3849 CrossRef CAS; (b) X. Yang, L. Zhao, T. Fox, Z.-X. Wang and H. Berke, Angew. Chem., Int. Ed., 2010, 49, 2058 CrossRef CAS; (c) M. Yang and F. Liu, J. Org. Chem., 2007, 72, 8969 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra21994h |
| This journal is © The Royal Society of Chemistry 2013 |