Poly(N-vinylimidazole) as an efficient catalyst for acetylation of alcohols, phenols, thiols and amines under solvent-free conditions
Nader
Ghaffari Khaligh
*
Department of Chemistry, College of Science, University of Guilan, Rasht, 41335-19141, Iran. E-mail: ngkhaligh@guilan.ac.ir; ngkhaligh@gmail.com; Fax: +982166934046; Tel: +982166431738
First published on 25th October 2012
Abstract
Poly(N-vinylimidazole) is able to promote instantaneous quantitative acetylation of a variety of functionalized alcohols, phenols, thiols and amines with acetic anhydride at room temperature under solvent-free conditions. This new method consistently has excellent yields and the catalyst can be reused and recovered several times. Furthermore, the reaction can even be carried out on a larger scale.
1. Introduction
Organic bases are catalysts for a wide range of reactions.1–5 The development of heterogeneous catalysts has become a major area of research in synthetic organic chemistry due to some potential advantages of these materials over homogeneous systems, such as simplified recovery, reusability, enhanced selectivity and reactivity, easy product isolation and incorporation in continuous reactors and microreactors.6The imidazole ring is a structural fragment of the side chain in most amino acids (i.e., histamine, histidine, etc.), which constitutes a part of almost all enzymes and is partly responsible for their catalytic activity. Imidazole is known to catalyze a number of biochemical reactions,7,8 therefore, the catalytic behaviors of monomeric9 and polymeric10 imidazole was extensively studied while simulating some biological processes.11,12 Imidazole-containing macromolecules catalyze the hydrolysis of p-nitrophenyl acetate,11 Michael addition of thiols to electron-deficient alkenes13 and aza-Michael reaction in water14 acting as base catalysts.
Poly(N-vinylimidazole) (PVIm) is a weak base (pKBH+ = 5–6).15 Its peculiar features in comparison with aliphatic amines are negligible protonation within the neutral pH range and a high capacity for hydrogen bonding.16,17 Poly(N-vinylimidazole) is known to form complexes with such metal ions as Cu(II),10 Zn(II),18 Cd(II),19 Ag(I)10 and Hg(II).20 The synthesized poly(N-vinylimidazole) with trimethoxysilyl terminal groups was chemically anchored (grafted) on magnetic nanoparticles that were to be used for the removal and recovery of heavy metals from industrial effluents.21a The practical applications of PVIm are numerous, ranging from dyestuffs, catalysts, corrosion inhibitors, ion exchange resins to their utility in quenching media and metal ion complexations.21b
It is worth noting that in several cases, the use of solvent-free conditions is necessary for the success of the process that can be dramatically slow or practically unfeasible using an organic solvent.22,23
The protection of alcohols, phenols, thiols and amines by the formation of esters and amides is one of the most important and widely used transformations in organic chemistry.24 The protection of such functional groups is often necessary during the course of various transformations in a synthetic sequence, especially in the construction of polyfunctional molecules, such as nucleosides, carbohydrates, steroids and natural products.25,26 A variety of procedures are routinely performed for the preparation of acetyl derivatives, including homogeneous or heterogeneous catalysts, such as DMAP and 4- pyrrolidinopyridine,27 TMEDA,28 Bu3P,29 iodine,30p-toluenesulfonic acid,31 alumina,32 montmorillonite K-10 and KSF,33 zeolite HSZ-360,34 zirconium sulfophenyl phosphonate,35 phosphomolybdic acid (PMA),36 acetonyltriphenylphosphonium bromide,37 Sc(OTf)3,38 trimethylsilyl trifluoromethanesulfonate (TMSOTf),39 Cu(OTf)2,40 copper(II) tetrafluoroborate,41a Mg(NTf2)2,41b In(OTf)3,42 Ce(OTf)3,43 silver triflate,44 magnesium bromide,45 bismuth(III) salts,46 Mg(ClO4)2,47a HClO4–SiO2,47b HBF4–SiO2,47c BiOClO4·xH2O,47d ferric perchlorate adsorbed on silica gel,48 zinc chloride,49 cobalt chloride,50 RuCl3,51 InCl3,52 ZrCl4,53 TaCl5,54 Cp2ZrCl255 and cerium polyoxometalate.56 However, some of the reported methods for the acetylation suffer from one or more of the disadvantages, such as drastic reaction conditions, expensive catalysts, hygroscopicity and thermal instability of the catalysts, use of halogen-containing solvents and catalysts, long reaction times and low yields. Therefore, the introduction of new methods and catalysts for the preparation of esters and amides is still in demand.
It is clear that green chemistry not only requires the use of environmentally benign reagents and solvents, but also the recovery and reuse of the catalyst. One way to overcome the problem of recyclability of the traditional reagents is to chemically anchor the reactive center onto a large surface area solid carrier.57 In these types of solids, the reactive centers are highly mobile, similar to homogeneous reagents, and at the same time these species have the advantage of being recyclable in the same fashion as heterogeneous reagents. Functional polymers have the potential advantages of small molecules with the same functional groups.58
Following our current interest based on the use of solid catalysts under solvent-free conditions,59 in this paper, we are describing our work on the successful use of non-toxic, environmentally benign and inexpensive poly(N-vinylimidazole) (PVIm) as a solid nucleophile catalyst for the acetylation of alcohols, phenols, thiols and amines with acetic anhydride at room temperature under solvent-free conditions. The novelty of this work is the use for the first time of PVIm for the acetylation of alcohols, phenols, thiols and amines with acetic anhydride. The results could be useful in finding new applications for this polymer.
2. Results and discussion
In continuation of our ongoing research program on the development of new catalysts and methods for organic transformations,59–61 here we wish to report the environmentally benign synthesis of poly(N-vinylimidazole) (PVIm) for acetylation of alcohols, phenols, thiols and amines at room temperature under solvent-free conditions (Scheme 1). PVIm was synthesized using a standard free-radical polymerization, as reported earlier.62,63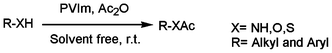 | ||
| Scheme 1 Acetylation of alcohols, phenols, thiols and amines at room temperature under solvent-free conditions. | ||
To begin with, a comparative study with imidazole (Im) and N-methylimidazole (MIm), instead of poly(N-vinylimidazole) (PVIm) was carried out. The acetylation of 4-chlorobenzyl alcohol by Ac2O with equivalent ratio was carried out in the presence of monomeric and polymeric acetylating agents at room temperature under solvent-free conditions (Table 1). As shown, after stirring for 6 h at room temperature in the absence of acetylating agents, the reaction did not proceed as monitored by TLC (Table 1, entry 1). On the other hand, in the presence of a certain amount of the Im, MIm and PVIm acetylating agents at room temperature, the reaction was completed with 100% conversion in 10, 5 and 5 min, respectively. When the acetylating agent was changed from imidazole to N-methylimidazole, the reaction time was improved (Table 1, entries 2, 3), however, when replacing N-methylimidazole with poly(N-vinylimidazole), no significant improvement was observed in the reaction time (Table 1, entries 3, 4). According to the results in Table 1, it can be concluded that both acetylating agents were effective for the acetylation of benzyl alcohol at room temperature, but PVIm is a heterogeneous catalyst, also as compared to N-methylimidazole and imidazole, poly(N-vinylimidazole) is highly stable and easy to handle.
| Entry | Catalyst | Amount (mg) | Time (min) | Yield (%)b |
|---|---|---|---|---|
| a Reaction conditions: substrate, 1.0 mmol; Ac2O, 1.0 mmol. b GC-MS yield. | ||||
| 1 | — | — | 6 h | N. R. |
| 2 | Imidazole | 50 | 10 | 92 |
| 3 | N-methylimidazole | 50 | 5 | 94 |
| 4 | Poly(N-vinylimidazole) | 50 | 5 | 94 |
In order to standardize the reaction, different amounts of PVIm were used to get 4-chlorobenzyl acetate from 4-chlorobenzyl alcohol and acetic anhydride at room temperature under solvent-free conditions, and the results are summarized in Table 2. It was clear that the reaction can be carried out in the presence of a catalytic amount of PVIm and the yields were excellent. It was found that the reaction was not possible without catalyst.
After these preliminary experiments, a catalytic amount of PVIm (20 mg) was used to convert a variety of functionalized alcohols, phenols, thiols and amines with acetic anhydride into respective esters and amides at room temperature under solvent-free conditions, and the results are presented in Table 3.
| Entry | Substrate | Product | Time (min) | Yield (%)b |
|---|---|---|---|---|
| a Reaction conditions: substrate, 1.0 mmol; Ac2O, 1.0 mmol; catalyst, 20 mg. b Isolated yield of the corresponding acetylated product. c Ac2O, 2.0 equiv.; isolated yield of the di-acetate. d Ac2O, 3.0 equiv.; isolated yield of the tri-acetate. | ||||
| 1 |
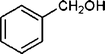
|
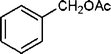
|
5 | 98 |
| 2 |
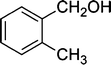
|
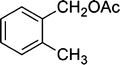
|
5 | 96 |
| 3 |
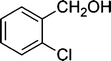
|
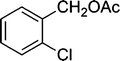
|
5 | 95 |
| 4 |
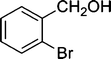
|
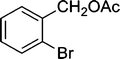
|
5 | 94 |
| 5 |
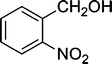
|
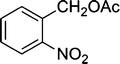
|
30 | 88 |
| 6 |
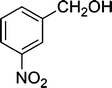
|
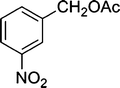
|
35 | 83 |
| 7 |
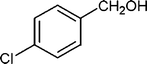
|
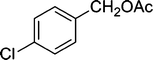
|
5 | 94 |
| 8 |
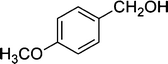
|
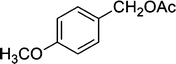
|
8 | 95 |
| 9 |
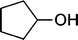
|
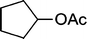
|
8 | 92 |
| 10 |
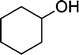
|
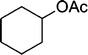
|
8 | 92 |
| 11 |
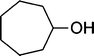
|
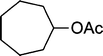
|
8 | 94 |
| 12 |
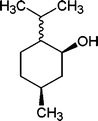
|
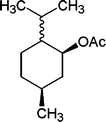
|
25 | 86 |
| 13 |
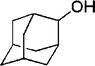
|
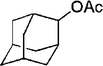
|
28 | 88 |
| 14 |
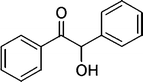
|
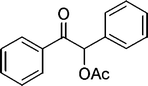
|
20 | 96 |
| 15 |
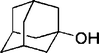
|
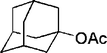
|
20 | 89 |
| 16 |
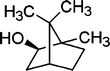
|
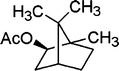
|
12 | 95 |
| 17 |
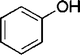
|
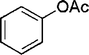
|
18 | 94 |
| 18 |
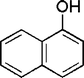
|
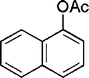
|
25 | 90 |
| 19 |
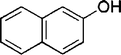
|
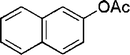
|
20 | 90 |
| 20 |
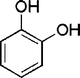
|
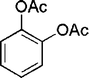
|
30 | 84c |
| 21 |
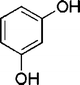
|
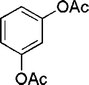
|
20 | 87c |
| 22 |
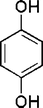
|

|
5 | 93c |
| 23 |
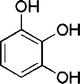
|
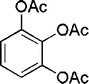
|
45 | 92d |
| 24 |
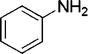
|
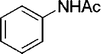
|
2 | 90 |
| 25 |
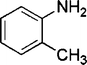
|
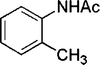
|
4 | 96 |
| 26 |
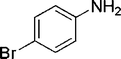
|
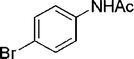
|
4 | 86 |
| 27 |
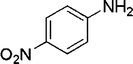
|
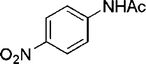
|
2 | 93 |
| 28 |
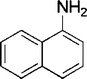
|
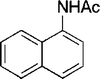
|
4 | 90 |
| 29 |
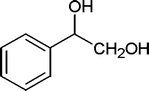
|
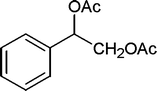
|
20 | 93c |
| 30 |
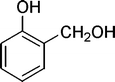
|
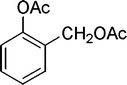
|
8 | 98c |
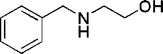
| 31 |
|
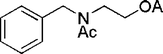
|
15 | 98c |
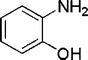
| 32 |
|
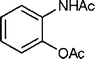
|
4 | 98c |
| 33 |
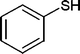
|
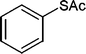
|
10 | 94 |
| 34 |
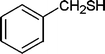
|
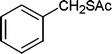
|
8 | 96 |
| 35 |
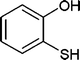
|
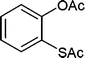
|
10 | 96c |
| 36 |
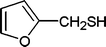
|
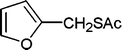
|
6 | 88 |
The results incorporated in Table 3 demonstrate the generality and scope of PVIm during the acetylation of structurally diverse alcohols and phenols. The reaction could be carried out with 1 equiv. of Ac2O at room temperature in 5–35 min. The compounds containing both electron withdrawing and electron donating groups reacted equally efficiently under the standard reaction conditions to give the acetylated products in excellent yields. The reaction conditions were mild enough not to induce any damage to moieties like the methoxyl group (Table 3, entry 9), which often underwent cleavage in the presence of strong acids or certain Lewis acids. Sterically hindered and electron deficient alcohols (Table 3, entries 5, 6 and 13–16) were efficiently acetylated under solvent-free conditions. Excellent selectivity was observed in that secondary and tertiary alcohols did not experience any competitive dehydration (Table 3, entries 9–16). In order to elucidate the mildness and stereospecificity of the PVIm catalyzed acetylation of alcohols, the reaction of optically active l(−)-Menthol and R(+)-Borneol were studied (Table 3, entries 12 and 16). We observed that l(−)-Menthol ([α]D= −49.0°, c = 10 in 95% ethanol, 99% e.e.) and R(+)-Borneol ([α]D= +37.9°, c = 5 in ethanol, 99% e.e.) react enantioselectivity with retention of the configuration on the benzylic center to provide l(−)-Menthyl acetate ([α]D = −73.0°, neat, 98% e.e.) and (+)-Bornyl acetate ([α]D = +41.5°, neat, 96% e.e.). The highest optical purity was obtained in an enantiopure form in high yield and under high regioselectivity using PVIm at room temperature under solvent-free conditions. The optical rotation of the product was determined and compared with that reported from Aldrich.
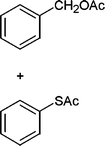
Also, we extended the use of PVIm for direct acetylation of amines (Table 3, entries 24–28) and thiols (Table 3, entries 33–36) with Ac2O. The superiority of PVIm was further established by the fact that direct acetylation of amines and thiols with 1.0 equiv. Ac2O could be achieved in 2–12 min at room temperature (86–96% yields) under the catalytic influence of 20 mg PVIm. Our protocol has some advantage because the two groups were acetylated during the reaction conditions (Table 3, entries 29–32, 35) and furyl mercaptane was transformed smoothly to the corresponding acetate derivative (Table 3, entry 36).
We have compared the obtained results in the acetylation of benzyl alcohol with acetic anhydride catalyzed by PVIm, with some heterogeneous and homogeneous catalysts, as reported in the literature (Table 4). It is clear that based on Table 4, some methods are superior in terms of reaction time, catalyst amount, or product yield. However, here we describe one or more of the limitations of the existing protocols, such as the use of halogenated catalysts, use of hazardous materials and use of costly catalysts (e.g. the triflates). Although the triflates, in general, are claimed as the most effective catalysts, they lead to competitive side reactions (e.g. dehydration and rearrangement etc.) for acid sensitive substrates.
| Entry | Catalyst | Solvent | Catalyst (mol%) | Time (min) | Yield (%)a | Ref. |
|---|---|---|---|---|---|---|
| a Isolated yield. b Temperature 60 °C. | ||||||
| 1 | I2 | Neat | 10 | 1 | 99 | 30 |
| 2 | montmorillonite KSF | Neat | 20 mg | 60 | 90 | 33 |
| 3 | zeolite HSZ-360 | Neat | 20 mg | 60 | 84 | 34 b |
| 4 | Cu(OTf)2 | CH2Cl2 | 2.5 | 30 | 97 | 40 |
| 5 | Ce(OTf)3 | CH3CN | 1 | 12 | 98 | 43 |
| 6 | Mg(ClO4)2 | Neat | 1 | 15 | 100 | 47 |
| 7 | CoCl2 | Neat | 0.5 | 240 | 98 | 50 |
| 8 | RuCl3 | CH3CN | 5 | 60 | 96 | 51 |
| 9 | InCl3 | Neat | 0.1 | 30 | 85 | 52 |
| 10 | Cp2ZrCl2 | Neat | 1 | 600 | 93 | 55 |
| 11 | PVIm | Neat | 20 mg | 5 | 98 | This work |
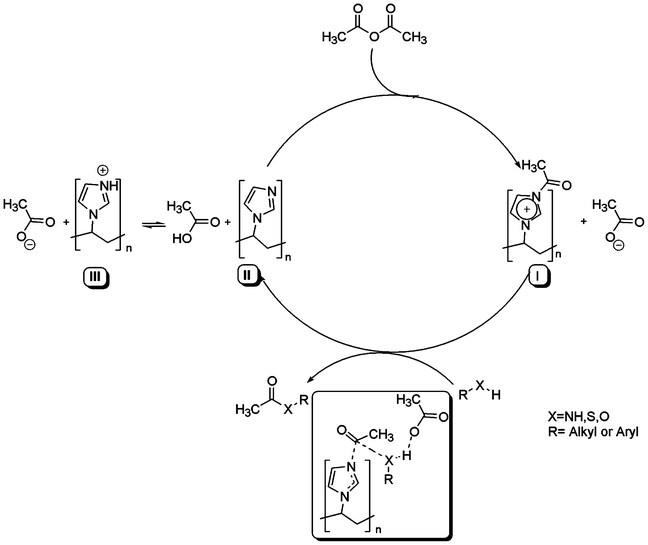
We are currently engaged in mechanistic studies to understand the precise role of PVIm. Although the actual mechanism of the reaction was not clear, based on the literature64–66 and FTIR spectra of PVIm (bottom) and the initial reaction of PVIm with acetic anhydride to generate an intermediate PVIm–Ac (top), the mechanism shown in Scheme 2 was selected as the most probable one.
 | ||
| Scheme 2 The plausible mechanism. | ||
The acetic anhydride-imidazole and acetic anhydride-N-methylimidazole systems proceed entirely by a nucleophilic and general base catalysis, respectively.66 It seems that the acetic anhydride-poly(N-vinylimidazole) system also reacts entirely via a nucleophilic route, with the intermediate formation of the acetylimidazolinium-anion ion-pair intermediate (I). This was followed by an essentially irreversible attack of an alcohol, phenol, thiol or amine to the intermediate (I) to generate an ester, thioester or amide product and to regenerate PVIm. PVIm can be used in high enough amounts to serve as both the proton scavenger (III) and the catalyst (I).
In the acetic anhydride-poly(N-vinylimidazole) system the spectral evidence for intermediate formation was seen (Fig. 1). PVIm (20 mg) was treated with Ac2O (1.0 mmol) in the absence of a substrate at room temperature under solvent-free conditions and magnetic stirring. After 5 min, the mixture was diluted with Et2O (25 ml) and the catalyst allowed to settle down. The supernatant ethereal solution was decanted off, the catalyst washed with Et2O (2 × 10 ml) and dried at room temperature under a vacuum for 2 h. The FTIR spectra of the acetylimidazolinium-anion ion-pair intermediate (top), was compared with FTIR spectra of PVIm (bottom).
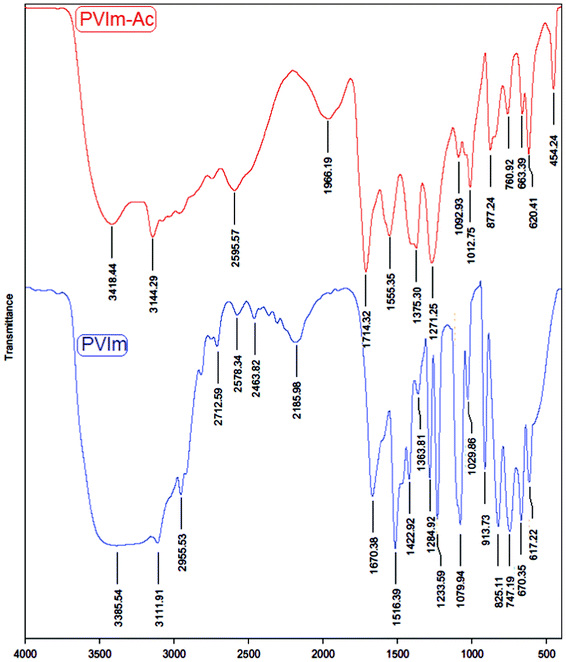 | ||
| Fig. 1 FTIR spectra for PVIm (bottom) and PVIm–Ac (top). | ||
The PVIm spectrum contains bands of stretching vibrations of imidazole rings (1516, 1422, 1285 and 1233 cm−1), stretching vibrations of azole C–H (1080 cm−1) and bending vibrations of heterocycles (914, 825 and 747 cm−1).67 The most important distinction between these two spectra was the development of a band at 1714 cm−1 following the functionalization of imidazole groups of PVIm with acetyl groups upon reaction with acetic anhydride. The imidazole units of PVIm corresponding to the ring deformation suffered a shift towards a higher wave number, (1013, 877, 761 cm−1) when the imidazole ring was acetylated with acetic anhydride. This interaction increased the stiffness of the associated ring and consequently more energy was required to deform the aromatic cycle, reflected in a higher wave number value. Additionally, the intensity of the band at about 1555 cm−1, as shown in Fig. 1, corresponded to positively charged nitrogen atoms. Combining the obvious and strong OH bands at 2595 and 1966 cm−1 in Fig. 1, we can infer that hydrogen bonding formed between the PVIm and acetic acid at equilibrium (Scheme 2). Furthermore, there was change of positions of the absorption peaks from 1670–1284 cm−1 for PVIm–Ac (top) in comparison with PVIm (bottom) in Fig. 1, which implies that the acetyl groups of PVIm–Ac were ionized.
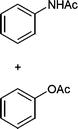
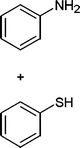
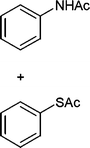
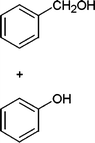
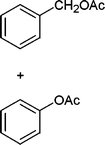
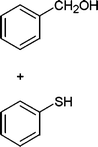
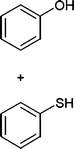
In addition, we also checked the chemoselectivity of this protocol. In Table 5, we demonstrate the inter-chemoselective acetylation of benzyl alcohol, phenol, thiophenol and aniline together. The results showed that aniline was acetylated selectively in the presence of benzyl alcohol, phenol and thiophenol (Table 5, entries 1, 2, 3). It was evident that only aniline was acetylated to acetanilide, while almost all of the starting benzyl alcohol, phenol and thiophenol were recovered at the end of the reaction. This may be considered as a useful practical achievement in the acetylation of amines in the presence of alcohols, phenols and thiols. Also benzyl alcohol was acetylated in the presence of phenol and thiophenol in 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15% and 84:16% yields, respectively (Table 5, entry 4, 5). Lastly, phenol was acetylated in the presence of thiophenol in 62:38% yield (Table 5, entry 6). On the other hand, the intra-chemoselective acetylation is shown in Table 3 (Entries 29–32). Results showed that the phenol, primary and secondary alcohol, and amine and aniline moiety were acetylated with together.
:15% and 84:16% yields, respectively (Table 5, entry 4, 5). Lastly, phenol was acetylated in the presence of thiophenol in 62:38% yield (Table 5, entry 6). On the other hand, the intra-chemoselective acetylation is shown in Table 3 (Entries 29–32). Results showed that the phenol, primary and secondary alcohol, and amine and aniline moiety were acetylated with together.

3. Experimental
3.1. General remarks
Chemicals were purchased from Merck, Aldrich and Fluka Chemical Companies and used without further purification. N-vinylimidazole was obtained from Aldrich Chemical Co. and was distilled under reduced pressure at 55 °C just prior to use. Azoisobutyronitrile (AIBN; BDH) (Fluka) was recrystallized from ethanol just before use. The purity determination of the products was accomplished by TLC on silica gel polygram SIL G/UV 254 plates. The MS were measured under GC (70 eV) conditions. The IR spectra were recorded on a Perkin Elmer 781 and Bruker Vector 22 Spectrophotometer. In all of the cases the 1H NMR spectra were recorded with a Bruker Avance 400 or 300 MHz instrument. Chemical shifts are reported in parts per million in CDCl3 with tetramethylsilane as an internal standard. 13C NMR data were collected on a Bruker Avance 100 or 75 MHz instrument.3.2. Typical procedure of acetylation
The substrate (alcohol, phenol, thiol or amine; 1.0 mmol) was treated with Ac2O (1.0 mmol) in the presence of PVIm (20 mg) at room temperature under solvent-free conditions and magnetic stirring. After completion of the reaction, as indicated by TLC, the mixture was diluted with Et2O (25 ml) and the catalyst allowed to settle down. The supernatant ethereal solution was decanted off, the catalyst washed with Et2O (2 ml) and the combined ethereal solution concentrated under a vacuum to afford the product identical (mp, IR, 1H and 13C NMR and GCMS) to an authentic sample of acetylated product. An important advantage of polymeric catalysts is that they can readily be separated from the products due to a considerable difference in the properties of high- and low-molecular compounds. As a result, such catalysts may be used repeatedly, which is profitable from the economic viewpoint and is environmentally benign. Thus, the use of poly(N-vinylimidazole) is promising from the viewpoint of “green chemistry”.68 The recovered catalyst was dried at 50 °C under a vacuum for 8 h. The recovered catalyst, after drying, was reused for four more consecutive acetylation reactions of benzyl alcohol (1.0 mmol) affording 98, 97, 97 and 96% yields in 2, 3, 3 and 5 min, respectively (Scheme 3).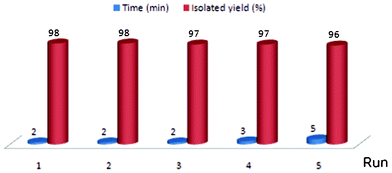 | ||
| Scheme 3 Recovery and reuse of the catalyst on the acetylation of benzyl alcohol with Ac2O at room temperature under solvent-free conditions. | ||
One of the difficulties of most of the heterogeneous basic catalysts arises from rapid poisoning by CO2 and H2O contained in the environment and the reactants. To be active catalysts, they need pretreatment at a specific temperature to remove adsorbed CO2 and H2O. To obtain full capabilities of PVIm, the reaction system should be kept free of impurities.
Notably, we have noticed that the conversion of benzyl alcohol to the respective acetate product can be carried out even on a larger scale (10 mmol) in 82% yield for 10 min without any difficulty using 20 mg of PVIm. Therefore, it indicates that a large-scale reaction was possible using the same amount of catalyst. It is important to point out that the present method was much cleaner and did not involve any chromatographic separation for a large-scale reaction. The facts that heterogeneous catalysts have good mechanical stability, can be easily handled as they are invariably low toxic, non-corrosive free flowing powder and are easily separated from the reaction mixture through filtration and reused, make them suitable for industrial applications.
3.3. The spectral data of some representative and new products
a) Table 2, entry 14: IR (neat) ν = 1738, 1697 cm−1; 1H NMR (CDCl3, 400 MHz): δ = 2.23 (s, 3H), 6.84 (s, 1H), 7.32–7.53 (m, 8H), 7.95 (d, J = 8.8 Hz, 2H); 13C (CDCl3, 100 MHz) δ = 20.1, 77.2, 128.2, 128.3, 128.6, 128.8, 133.1, 133.1, 134.1, 169.9, 193.3 ppm.b) Table 2, entry 16: IR (neat) ν = 1726 cm−1; [α]25 = −38° (neat); 1H NMR (CDCl3, 400 MHz): δ = 0.82 (s, 3H), 0.88 (s, 3H), 0.92 (s, 3H), 1.26 (m, 4H), 1.72 (m, 2H), 2.08 (s, 3H), 2.36 (m, 1H), 4.89 (d, J = 9.8 Hz, 1H); 13C (CDCl3, 100 MHz) δ = 13.4, 18.7, 19.6, 21.2, 27.0, 28.0, 36.7, 44.8, 47.7, 48.6, 79.8, 171.3 ppm.
c) Table 2, entry 19: mp 70–72 °C; IR (KBr) ν = 1756 cm−1; 1H NMR (CDCl3, 400 MHz): δ = 2.38 (s, 3H), 7.25 (d, J = 8.8 Hz, 1H), 7.46 (m, 2H), 7.55 (s, 1H), 7.79 (m, 3H); 13C NMR (CDCl3, 100 MHz) δ = 21.2, 118.5, 121.1, 125.7, 126.5, 127.6, 127.7, 129.4, 131.4, 133.7,148.3, 169.6 ppm.
d) Table 2, entry 23: IR (neat) ν = 1768 cm−1; 1H NMR (CDCl3, 400 MHz): δ = 2.31 (s, 6H), 2.31 (s, 3H), 7.14 (d, J = 8.0 Hz, 2H), 7.28 (dd, J = 8.8 and 7.6 Hz, 1H) ppm; 13C NMR (CDCl3, 100 MHz): δ = 20.2, 20.7, 120.7, 125.9, 134.7, 143.6, 167.0, 167.9 ppm; GCMS: m/z = 252 [M+].
e) Table 2, entry 28: IR (neat) ν: 3295, 1635 cm−1; 1H NMR (CDCl3): δ = 2.30 (s, 3H), 7.45 (d, J = 7.6 Hz, 2H), 7.48–7.87 (m, 6H) ppm; 13C NMR (CDCl3, 100 MHz): δ = 22.8, 118.5, 122.3, 122.8, 125.2, 126.1, 127.3, 127.8, 128.4, 128.5, 133.1, 169.9 ppm.
f) Table 2, entry 32: IR (neat) ν = 3320, 1730, 1682 cm−1; 1H NMR (CDCl3, 400 MHz): δ = 2.16 (s, 3H), 2.36 (s, 3H), 7.14–7.17 (m, 2H), 7.22–7.25 (m, 1H), 7.39 (br s, 1H), 8.11 (d, J = 7.6 Hz, 1H) ppm; 13C NMR (CDCl3, 100 MHz): δ = 20.6, 22.7, 117.8, 120.7, 123.9, 125.9, 130.7, 140.5, 167.0, 167.9 ppm.
h) Table 2, entry 35: IR (neat) ν = 1765, 1679 cm−1; 1H NMR (400 MHz , CDCl3): δ = 2.33 (s, 3H), 2.45 (s, 3H), 7.24 (d, J = 8.0 Hz, 1H), 7.33 (t, J = 7.6 Hz, 1H), 7.50 (t, J = 7.6 Hz, 1H) 7.53 (d, J = 7.6 Hz, 1H) ppm; 13C-NMR (100 MHz, CDCl3): δ = 20.7, 26.2, 120.7, 125.9, 130.2, 134.7, 143.5, 167.0, 171.9 ppm.
i) Table 2, entry 36: IR (neat) ν = 1696 cm−1; 1H NMR (400 MHz, CDCl3): δ = 2.36 (s, 3H), 4.29 (s, 2H), 6.01 (d, J = 2.8 Hz, 1H), 6.23 (dd, J = 2.8 and 0.8 Hz, 1H), 7.22 (d, J = 0.8, 1H) ppm; 13C-NMR (100 MHz, CDCl3): δ = 22.4, 29.7, 107.2, 110.7, 141.7, 152.7, 194.3 ppm.
4. Conclusions
PVIm is a new and highly efficient catalyst for acetylation of alcohols, phenols, thiols and amines. The low cost, ease of handling and, with the increasing environmental concern,69 the solvent-free conditions employed in the present method make it “environmentally friendly” and therefore useful for industrial applications. The methodology of chemical efficiency, simplified experimental procedures and the minimization of the amount of organic solvent used which is to facilitate the separation of the product from the heterogeneous catalyst.References
- S. Bertelsen and K. A. Jørgensen, Chem. Soc. Rev., 2009, 38, 2178–2189 RSC.
- D. W. C. McMillan, Nature, 2008, 45, 304–308 Search PubMed.
- A. Dondoni and A. Massi, Angew. Chem., Int. Ed., 2008, 47, 4638–4660 CrossRef.
- P. Melchiorre, M. Merigo, A. Carlone and G. Bartoli, Angew. Chem., Int. Ed., 2008, 47, 6138–6171 CrossRef CAS.
- C. F. Barbas, Angew. Chem., Int. Ed., 2008, 47, 42–47 CrossRef.
- (a) ed., P. I. Dalko, Enantioselective Organocatalysis, Wiley-VCH, Weinheim, 2007 Search PubMed; (b) A. J. Fatiadi, Synthesis, 1987, 85–127 CrossRef CAS; (c) F. M. Menger and C. Lee, J. Org. Chem., 1979, 44, 3446–3448 CrossRef CAS; (d) K. Smith, Solid Supports and Catalysis in Organic Synthesis, Ellis Horwood and PTR Prentice Hall, New York ( 1992) Search PubMed; (e) P. Laszlo, Comprehensive Organic Synthesis, Vol. 7, ed.: Trost, B. M., New York, Pergamon, 1991, p 839 Search PubMed; (f) ed., P.I. Dalko, Enantioselective Organocatalysis,Wiley-VCH, Weinheim, 2007 Search PubMed.
- I. V. Berezin and K. Martinek, Osnovy fizicheskoi khimii fermentativnogo kataliza [Foundations of the Physical Chemistry of Enzymatic Catalysis], Vysshaya shkola, Moscow, 1977, 280pp, (in Russian) Search PubMed.
- M. L. Bender, R. J. Bergeron and M. Komiyama, The Bioorganic Chemistry of Enzymatic Catalysis, New York, Wiley, 1984. Translated under the title Bioorganicheskaya khimiya fermentativnogo kataliza, Moscow, Mir, 1987, p. 352 Search PubMed.
- (a) J. F. Kirsch and W. P. Jencks of Enzymatic, Catalysis, Moscow: Vysshaya Shkola, 1977, 280 Search PubMed; (b) J. F. Kirsch and W. P. Jencks, J. Am. Chem. Soc., 1964, 86, 833–837 CrossRef CAS.
- D. H. Gold and H. P. Gregor, J. Phys. Chem., 1960, 64, 1461–1463 CAS.
- I. M. Okhapkin, L. M. Bronstein, E. E. Makhaeva, V. G. Matveeva, E. M. Sulman, M. G. Sulman and A. R. Khokhlov, Macromolecules, 2004, 37, 7879–7883 Search PubMed.
- V. L. Lozinsky, I. A. Simenel, V. K. Kulakova, E. A. Kurskaya, T. A. Babushkina, T. P. Klimova, T. V. Burova, A. S. Dubovik, V. Y. Grinberg, I. Y. Galaev, B. Mattiasson and A. R. Khokhlov, Macromolecules, 2003, 36, 7308–7323 Search PubMed.
- I. P. Beletskaya, E. A. Tarasenko, A. R. Khokhlov and V. S. Tyurin, Russ. J. Org. Chem., 2007, 43, 1733–1736 Search PubMed.
- I. P. Beletskaya, E. A. Tarasenko, A. R. Khokhlov and V. S. Tyurin, Russ. J. Org. Chem., 2010, 46, 461–467 Search PubMed.
- N. L. Mazyar, V. V. Annenkov, V. A. Kruglova, S. M. Ananiev, E. N. Danilovtseva, A. V. Rokhin and S. V. Zinchenko, Russ. Chem. Bull., 2000, 49, 2013–2017 Search PubMed.
- N. L. Mazyar, V. V. Annenkov, V. A. Kruglova, D. C. D. Toriachinova and E. N. Danilovtseva, Vysokomol. Soedin. A., 1999, 41, 357–362 Search PubMed.
- V. V. Annenkov, E. A. Filina, E. N. Danilovtseva, S. V. Zinchenko and A. I. Michaleva, J. Sol-Gel Sci. Technol., 2003, 27, 163–166 Search PubMed.
- K. J. Liu and H. P. Gregor, J. Phys. Chem., 1965, 69, 1252–1259 CrossRef CAS.
- C. Tanford and M. L. Wagner, J. Am. Chem. Soc., 1953, 75, 434–435 Search PubMed.
- M. J. Molina, M. R. Gomez-Anton, B. L. Rivas, H. A. Maturana and I. F. Pierola, J. Appl. Polym. Sci., 2001, 79, 1467–1475 Search PubMed.
- (a) M. Takafuji, S. Ide, H. Ihara and Z. Xu, Chem. Mater., 2004, 16, 1977–1983 CrossRef CAS; (b) R. C. Sutton, L. Thai, J. M. Hewitt, C. L. Voycheck and J. S. Tan, Macromolecules, 1988, 21, 2432–2439 CrossRef CAS.
- T. Angelini, F. Fringuelli, D. Lanari, F. Pizzo and L. Vaccaro, Tetrahedron Lett., 2010, 51, 1566–1569 CrossRef CAS.
- F. Fringuelli, D. Lanari, F. Pizzo and L. Vaccaro, Curr. Org. Synth., 2009, 6, 203–208 Search PubMed.
- (a) J. Otera, Esterification: Methods, Reactions and Applications, 1st ed., Wiley–VCH, 2003 Search PubMed; (b) T. W. Greene and P. G. M. Wutz, Protective Groups in Organic Synthesis, 3rd edn., Wiley, New York, 1999 Search PubMed.
- X. L. Sun, T. Kai, H. Takayanagi and K. Furuhata, Synlett, 1999, 9, 1399–1341 Search PubMed.
- J. S. Yadav, A. V. Narsaiah, A. K. Basak, P. R. Goud, D. Sreenu and K. Nagaiah, J. Mol. Catal. A: Chem., 2006, 255, 78–80 CrossRef CAS.
- (a) E. F. V. Scriven, Chem. Soc. Rev., 1983, 12, 129–161 RSC; (b) G. Höfle, V. Steglich and H. Vorbrüggen, Angew. Chem., Int. Ed. Engl., 1978, 17, 569–583 CrossRef; (c) A. Sakakura, K. Kawajiri, T. Ohkubo, Y. Kosugi and K. Ishihara, J. Am. Chem. Soc., 2007, 129, 14775–14779 CrossRef CAS.
- T. Sano, K. Ohashi and T. Oriyama, Synthesis, 1999, 1141–1144 CrossRef CAS.
- (a) E. Vedejs, N. S. Bennett, L. M. Conn, S. T. Diver, M. Gingras, S. Lin, P. A. Oliver and M. J. Peterson, J. Org. Chem., 1993, 58, 7286–7288 CrossRef CAS; (b) E. Vedjes and T. S. Diver, J. Am. Chem. Soc., 1993, 115, 3358–3359 CrossRef CAS.
- (a) R. Borah, N. Deka and J. Sarma, J. Chem. Res. (S), 1997, 110–111 RSC; (b) P. Phukan, Tetrahedron Lett., 2004, 45, 4785–4787 CrossRef CAS.
- A. C. Cope and E. C. Herrich, Organic Synthesis Collective, vol. IV, Wiley, New York, 1963, p. 304 Search PubMed.
- (a) S. S. Rana, J. J. Barlow and K. L. Matta, Tetrahedron Lett., 1981, 22, 5007–5010 CrossRef CAS; (b) G. W. Breton, M. J. Kurtz and S. L. Kurtz, Tetrahedron Lett., 1997, 38, 3825–3828 CrossRef CAS.
- (a) A. X. Li, T. S. Li and T. H. Ding, Chem. Commun., 1997, 1389–1390 RSC; (b) H. Hagiwara, K. Morohashi, T. Suzuki, M. Ando, I. Yamamoto and M. Kato, Synth. Commun., 1998, 28, 2001–2006 CAS.
- R. Ballini, G. Bosica, S. Carloni, L. Ciaralli, R. Maggi and G. Sartori, Tetrahedron Lett., 1998, 39, 6049–6052 CrossRef CAS.
- M. Curini, F. Epifano, M. C. Marcotullio, O. Rosati and M. Rossi, Synth. Commun., 2000, 30, 1319–1329 CAS.
- S. T. Kadam and S. S. Kim, Synthesis, 2008, 267–268 CAS.
- A. T. Khan, L. H. Choudry and S. Ghosh, Eur. J. Org. Chem., 2005, 2782–2787 CrossRef CAS.
- K. Ishihara, M. Kubota, H. Kurihara and H. Yamamoto, J. Org. Chem., 1996, 61, 4560–4567 CrossRef CAS.
- (a) P. A. Procopiou, S. P. D. Baugh, S. S. Flack and G. G. A. Inglis, Chem. Commun., 1996, 2625–2626 RSC; (b) P. A. Procopiou, S. P. D. Baugh, S. S. Flack and G. G. A. Inglis, J. Org. Chem., 1998, 63, 2342–2347 CrossRef CAS.
- (a) P. Saravanan and V. K. Singh, Tetrahedron Lett., 1999, 40, 2611–2614 CrossRef CAS; (b) K. L. Chandra, P. Saravan, R. K. Singh and V. K. Singh, Tetrahedron, 2002, 58, 1369–1374 CrossRef.
- (a) A. K. Chakraborti, R. Gulhane and Shivani, Synthesis, 2004, 111–115 CrossRef CAS; (b) A. K. Chakraborti and Shivani, J. Org. Chem., 2006, 71, 5785–5788 CrossRef CAS.
- K. K. Chauhan, C. G. Frost, I. Love and D. Waite, Synlett, 1999, 1743–1744 CrossRef CAS.
- R. Dalpozzo, A. D. Nino, L. Maiuolo, A. Procopiou, M. Nardi, G. Bartoli and R. Romeo, Tetrahedron Lett., 2003, 44, 5621–5624 CrossRef CAS.
- R. Das and D. Chakraborty, Synthesis, 2011, 1621–1625 CAS.
- S. V. Pansare, M. G. Malusare and A. N. Rai, Synth. Commun., 2000, 30, 2587–2592 CAS.
- I. Mohammadpoor-Baltork, H. Aliyan and A. R. Khosropour, Tetrahedron, 2001, 57, 5851–5854 CrossRef CAS.
- (a) A. K. Chakraborti, L. Sharma, R. Gulhane and Shivani, Tetrahedron, 2003, 59, 7661–7668 CrossRef CAS; (b) A. K. Chakraborti and R. Gulhane, Chem. Commun., 2003, 1896–1897 RSC; (c) A. K. Chakraborti and R. Gulhane, Tetrahedron Lett., 2003, 44, 3521–3525 CrossRef CAS; (d) A. K. Chakraborti, R. Gulhane and Shivani, Synlett, 2003, 12, 1805–1808 Search PubMed.
- A. Parmar, J. Kaur, R. Goyal, B. Kumar and H. Kumar, Synth. Commun., 1998, 28, 2821–2826 CAS.
- R. H. Baker and F. G. Bordwell, Organic Synthesis Collective, vol. III, Wiley, New York, 1995, p. 141 Search PubMed.
- J. Iqbal and R. R. Srivastava, J. Org. Chem., 1992, 57, 2001–2007 CrossRef CAS.
- S. K. De, Tetrahedron Lett., 2004, 45, 2919–2922 CrossRef.
- A. K. Chakraborti and R. Gulhane, Tetrahedron Lett., 2003, 44, 6749–6753 CrossRef CAS.
- A. K. Chakraborti and R. Gulhane, Synlett, 2004, 627–630 CrossRef CAS.
- S. Chandrasekhar, T. Ramachander and M. Takhi, Tetrahedron Lett., 1998, 39, 3263–3266 CrossRef CAS.
- M. L. Kantam, K. Aziz and P. R. Likhar, Catal. Commun., 2006, 7, 484–487 CrossRef CAS.
- V. Mirkhani, S. Tangestaninejad, M. Moghadam, B. Yadollahi and L. Alipanah, Monatsh. Chem., 2004, 135, 1257–1263 CrossRef CAS.
- J. A. Melero, R. van Grieken and G. Morales, Chem. Rev., 2006, 106, 3790–3812 CrossRef CAS.
- (a) A. Akelah and D. C. Sherindton, Chem. Rev., 1981, 81, 557–587 CrossRef CAS; (b) D. C. Sherington and P. Hodge, Synthesis and Separation Using Functional Polymers, Wiley, Chichester ( 1988) Search PubMed; (c) K. Takemoto, Y. Inaki and R. M. Ottenbrite, Functional Monomers and Polymers, Dekker, New York, 1987 Search PubMed; (d) S. V. Ley, I. R. Baxendale, R. N. Bream, P. S. Jackson, A. G. Leach, D. A. Longbottom, M. Nesi, J. S. Scott, R. I. Storer and S. J. Taylor, J. Chem. Soc., Perkin Trans. 1, 2000, 1, 3815–4195 Search PubMed.
- F. Shirini and N. G. Khaligh, J. Iran. Chem. Soc., 2012, 9, 495–502 Search PubMed.
- (a) N. G. Khaligh, J. Mol. Catal. A: Chem., 2011, 349, 63–70 CrossRef CAS; (b) N. G. Khaligh and F. Shirini, J. Mol. Catal. A: Chem., 2011, 348, 20–29 CrossRef CAS; (c) F. Shirini and N. G. Khaligh, Phosphorus, Sulfur Silicon Relat. Elem., 2011, 186, 2156–2165 Search PubMed.
- (a) N. G. Khaligh, Tetrahedron Lett., 2012, 53, 1637–1640 CrossRef CAS; (b) N. G. Khaligh, Ultrason. Sonochem., 2012, 19, 736–739 CrossRef CAS; (c) N. G. Khaligh, RSC Adv., 2012, 2, 3321–3327 RSC; (d) F. Shirini and N. G. Khaligh, Monatsh. Chem., 2012, 143, 631–635 Search PubMed.
- A. Chapiro and Z. Mankowski, Eur. Polym. J., 1988, 24, 1019–1028 Search PubMed.
- B. Cabot, A. Deratani and A. Foissy, Colloids Surf. A Physicochem. Eng. Aspects, 1998, 139287–297 Search PubMed.
- (a) S. Xu, I. Held, B. Kempf, H. Mayr, W. Steglich and H. Zipse, Chem. Eur. J., 2005, 11, 4751–4757 CrossRef CAS; (b) C. B. Fischer, S. Xu and H. Zipse, Chem. Eur. J., 2006, 12, 5779–5784 CrossRef CAS.
- (a) T. L. Gilchrist, in Heterocyclic Chemistry, 3rd edn., Longman, Singapore, 1997, Chapter 8 Search PubMed; (b) M. R. Grimmett, in Advances in Heterocyclic Chemistry, ed.,A. R. Katritzky and A. J. Boulton, Academic Press, New York, 27, 1980 Search PubMed.
- (a) K. A. Connors and N. K. Pandit, Anal. Chem., 1978, 50, 1542–1545 CrossRef CAS; (b) N. K. Pandit and K. A. Connors, J. Pharm. Sci., 1982, 71, 485–491 CAS.
- J. L. Lippert, J. A. Robertson, J. R. Havens and J. S. Tan, Macromolecules, 1985, 18, 63–67 CrossRef CAS.
- R. A. Sheldon, I. W. C. E. Arends and U. Hanefeld, Green Chemistry and Catalysis, Weinheim, Wiley, 2007 Search PubMed.
- R. L. Garrett, in Designing Safer Chemicals, ed. R. L. Garrett and S. C. De Vito, American Chemical Society Symposium Series 640, Washington DC, 1996, ch. 1 Search PubMed.
| This journal is © The Royal Society of Chemistry 2013 |