Tunable gold nanostructures with nanocapsules as template reaction vessels
Ramjee
Balasubramanian
*a,
Srujana
Prayakarao
a,
Sangbum
Han
a and
Wei
Cao
b
aDepartment of Chemistry and Biochemistry, 4541 Hampton Blvd., Old Dominion University, Norfolk, VA, USA. E-mail: bramjee@odu.edu; Tel: +1 757-683-3039
bApplied Research Center, Old Dominion University, Newport News, VA, USA
First published on 5th October 2012
Abstract
Hollow, polymeric, thiol-ene resorcinarene nanocapsules can act as template reaction vessels for the synthesis of gold nanoshells or spherical aggregates of gold nanoparticles depending on the reaction conditions. The spherical nanoparticle aggregates were evaluated as catalysts in the biphasic reduction of 4-nitrophenol.
Templates provide a convenient route for the synthesis and/or self-assembly of plasmonic nanostructures.1–4 Among various hard and soft templates, polymers offer numerous advantages such as ready availability, precise functionalization protocols, well-defined binding sites, self-organization etc.1,2,5 One of the major challenges in the fabrication of polymer nanoparticle composites is the effective dispersion of nanoparticles in polymer hosts,6 often requiring modification of either component.5 Though most of the work has focused on block copolymers, other polymeric platforms have also been integrated with nanoparticles.1 Among them, hollow polymeric capsules are attractive candidates due to their inherent guest encapsulation and release capabilities.7,8 When compared to the extensively studied assembly of nanostructures on macroscopically flat surfaces, the template assembly of nanostructures on curved, three-dimensional substrates is still nascent.4,9 The fabrication of nanocapsule–nanoparticle ensembles typically involves the embedding of the nanoparticles at a very early stage.10,11 Direct synthesis of nanoparticles inside container molecules has also been reported, but they lead to low loading of nanoparticles.12,13 Hence, there exists a need to develop simple, tunable, modular approaches for the preparation of controlled hybrid architectures.
The present work employs a hollow, polymeric resorcinarene nanocapsule as a template reaction vessel for the synthesis and self-assembly of gold nanostructures. We have recently reported a direct, template-free approach for the fabrication of resorcinarene nanocapsules by the photopolymerization of a thiol-ene monomer, resorcinarene tetra alkene tetra thiol (RTATT, Scheme 1), in chloroform.14,15 We envisaged that the RTATT nanocapsules containing ubiquitous sulfide linkages and thiol functional groups can act as multivalent templates for the synthesis and assembly of metallic nanostructures. In principle, nanocapsules as templates could stabilize nanoparticles both inside and outside the polymer shell in a number of distinct ways. In this communication, we show that the nature and shape of the gold nanostructure formed with the RTATT nanocapsule templates, i.e., nanoshells or aggregates of smaller individual nanoparticles is a function of reaction conditions (Scheme 1).
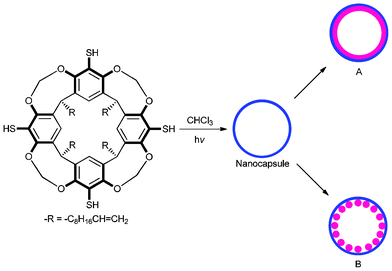 | ||
| Scheme 1 Resorcinarene nanocapsules as template reaction vessels for tunable gold nanostructures. | ||
Brust's two-phase method16 was employed for the synthesis of the gold nanostructures with resorcinarene nanocapsule templates. Briefly, a mixture of TOA+AuCl4− [obtained by the phase transfer of HAuCl4 (16.6 mg, 0.049 mmol) with tetraoctylammonium bromide (TOABr, 77.3 mg, 0.141 mmol)] and RTATT nanocapsules (prepared from 0.006 mmol of monomer, Fig. 1a) in chloroform were reduced with aqueous sodium borohydride (24 mg, 0.634 mmol) at 0 °C for 1 h. The TEM analysis of the product showed both smaller individual gold nanoparticles and much larger nanostructures (Fig. 1b). A closer look at the latter (Fig. 1c–1g) showed formation of a gold nanoshell with a darker rim (Fig. 1h) and a lighter core (Fig. 2). Our attempts to visualize the surrounding nanocapsule by OsO4 staining (Fig. 1, images d–f) were unsuccessful. The HRTEM image (Fig. 2) of the center of the nanoshell showed the presence of several single crystalline regions with varying grain sizes and shapes as indicated by representative white geometric shapes in Fig. 2. They also showed the presence of Moiré fringes (representative ones are shown in Fig. 2 in grey circles), which usually arise from the interference of two sets of crystalline planes with common periodicities. The TEM images (Fig. 1 and 2) and the presence of Moiré fringes support the formation of gold nanostructures somewhat similar to structure A in Scheme 1. Interestingly, these nanoshells were often decorated with smaller spherical nanoparticle satellites (Fig. 1c and 1e). The thickness of the nanoshells formed was typically ∼12 nm (Fig. 1h). In certain instances, gold nanoshell formation was interrupted as indicated by an arrow in Fig. 1f and 1g. This could perhaps be due to a ruptured wall in the parent nanocapsule.17
 | ||
| Fig. 1 TEM analysis of the gold nanoshells (b–h) prepared with RTATT nanocapsules (a, OsO4 stained sample). | ||
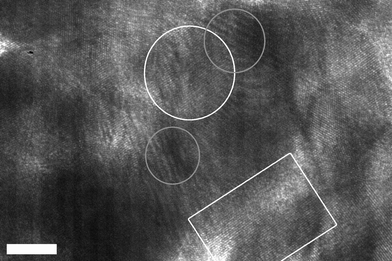 | ||
| Fig. 2 HRTEM analysis (scale bar = 5 nm) of the center of a gold nanoshell showing representative single-crystalline regions outlined in white and Moiré patterns outlined in grey. | ||
The selected area electron diffraction (SAED) of a gold nanoshell showed well crystallized gold within the nanoshell (Fig. 3a). Characteristic diffraction rings for polycrystalline gold were observed and indexed as (111), (200), (220), (311), and (400) crystalline facets. Dynamic light scattering (DLS) analysis of these nanoshells gave a hydrodynamic diameter of ∼141 nm (Fig. 3b). Not surprisingly, the extinction spectra of the as prepared nanoshells (NC–Au NS) showed a peak at 531 nm with a substantial tail in the NIR region (Fig. 3c). Most of the individual nanoparticles could be separated from the gold nanoshells by allowing the latter to preferentially sediment overnight (Fig. 3c). The sedimented nanoshells could be readily redispersed in chloroform or an RTATT solution in chloroform by gentle shaking. The extinction spectra of the nanoshells redispersed in chloroform (NC–Au NS ppt) showed a peak at ∼547 nm with a uniformly rising slope from 1200 nm (Fig. 3c). Though the extinction spectra of these nanoparticle decorated nanoshell architectures are distinct from other nanoshells,18–22 it is somewhat similar to the recently reported gold lace nanoshells23 or gold nanorattles.24 The observed extinction spectrum could be due to the interaction of the smaller nanoparticle satellites (Fig. 1c and 1e) with the nanoshell23 and/or the entrapment of spherical nanoparticles inside these nanoshells.24 The atomic force microscopy (AFM) analysis (Fig. 3d) of such redispersed nanoshells gave an average size of 130 ± 12 nm, which was in good agreement with the DLS data and the dimensions of the parent nanocapsules.
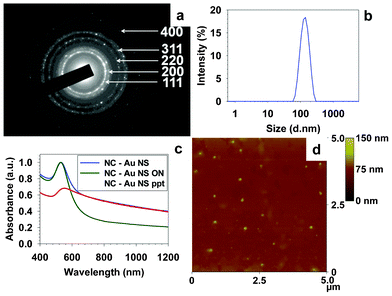 | ||
| Fig. 3 SAED (a), DLS (b), and AFM (d) analysis of the gold nanoshells prepared with RTATT nanocapsules. (c) UV-vis spectra of as prepared nanoshells containing individual nanoparticles (NC–Au NS), the supernatant obtained after overnight sedimentation (NC–Au NS ON) and the redispersed precipitate (NC–Au NS ppt) in chloroform. | ||
Notably, the concentration of the metal salt played a crucial role in the formation of the nanoshells. Upon increasing the S![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Au ratio from 0.5 to 0.8 (by varying the amount of HAuCl4 while keeping the RTATT concentration constant), spherical aggregates of gold nanoparticles were obtained (Fig. 4 a–e). This is somewhat similar to the observation of Murray and coworkers, who have shown that a higher thiol to Au ratio leads to the formation of gold nanoparticles with smaller core sizes.25 A closer look at these aggregates clearly revealed closely packed bilayer stacking of individual gold nanoparticles (Fig. 4 b–e), appearing somewhat similar to structure B in Scheme 1. A distinctive feature of this in situ nanoparticle synthesis and template assembly is that the size of the individual nanoparticles comprising the aggregates was significantly larger than the individual nanoparticles present outside (Fig. 4 b–e). It is possible that for the nanoparticles nucleating and growing inside the nanocapsules, there may only be partial/incomplete passivation from the nanocapsule, resulting in the formation of larger nanoparticles. Occasionally, the TEM image (Fig. 4e) also showed the presence of an organic material encapsulating the nanoparticle aggregates. Such spherically assembled gold nanoparticle ensembles were not observed when TOA+AuCl4− was reduced in the presence of RTATT monomer (Fig. 4f). The extinction spectra of the nanoparticles prepared in the presence of RTATT nanocapsules and monomer showed significant differences. When compared to the surface plasmon resonance (SPR) of the individual gold nanoparticles (RTATT-Au NP) at 526 nm, the as prepared gold nanoparticle aggregates (NC–Au NP) had a broadened SPR centred around 538 nm (Fig. 4g). As before, the resorcinarene nanocapsule encapsulated gold nanoparticle aggregates could be readily separated from the individual nanoparticles by allowing the former to sediment preferentially overnight. The sedimented particles could be readily redispersed in chloroform by gentle shaking, and their extinction spectra (NC–Au NP ppt, Fig. 4g) showed a significantly red shifted and broadened SPR ∼570 nm. Such broadening could be attributed to plasmonic coupling between the assembled nanoparticles.26
:Au ratio from 0.5 to 0.8 (by varying the amount of HAuCl4 while keeping the RTATT concentration constant), spherical aggregates of gold nanoparticles were obtained (Fig. 4 a–e). This is somewhat similar to the observation of Murray and coworkers, who have shown that a higher thiol to Au ratio leads to the formation of gold nanoparticles with smaller core sizes.25 A closer look at these aggregates clearly revealed closely packed bilayer stacking of individual gold nanoparticles (Fig. 4 b–e), appearing somewhat similar to structure B in Scheme 1. A distinctive feature of this in situ nanoparticle synthesis and template assembly is that the size of the individual nanoparticles comprising the aggregates was significantly larger than the individual nanoparticles present outside (Fig. 4 b–e). It is possible that for the nanoparticles nucleating and growing inside the nanocapsules, there may only be partial/incomplete passivation from the nanocapsule, resulting in the formation of larger nanoparticles. Occasionally, the TEM image (Fig. 4e) also showed the presence of an organic material encapsulating the nanoparticle aggregates. Such spherically assembled gold nanoparticle ensembles were not observed when TOA+AuCl4− was reduced in the presence of RTATT monomer (Fig. 4f). The extinction spectra of the nanoparticles prepared in the presence of RTATT nanocapsules and monomer showed significant differences. When compared to the surface plasmon resonance (SPR) of the individual gold nanoparticles (RTATT-Au NP) at 526 nm, the as prepared gold nanoparticle aggregates (NC–Au NP) had a broadened SPR centred around 538 nm (Fig. 4g). As before, the resorcinarene nanocapsule encapsulated gold nanoparticle aggregates could be readily separated from the individual nanoparticles by allowing the former to sediment preferentially overnight. The sedimented particles could be readily redispersed in chloroform by gentle shaking, and their extinction spectra (NC–Au NP ppt, Fig. 4g) showed a significantly red shifted and broadened SPR ∼570 nm. Such broadening could be attributed to plasmonic coupling between the assembled nanoparticles.26
 | ||
| Fig. 4 TEM (a–e) and UV-vis (g, NC–Au NP) of gold nanoparticle aggregates prepared with RTATT nanocapsules. TEM (f) and UV-vis (g, RTATT-Au NP) of gold nanoparticles prepared with RTATT monomer. | ||
We tested the nanocapsule templated gold nanoparticle aggregates (NC–Au NP) as catalysts in the reduction of para-nitrophenol with sodium borohydride. A variety of metallic nanoparticle catalysts,27,28 including core-shell architectures,29,30 have been shown to be effective in carrying out this reduction. We employed a bi-phasic approach,28 where the nanoparticle aggregates were present in the organic phase (chloroform) and 4-nitrophenol and sodium borohydride were present in the aqueous phase. In a typical catalytic reaction, to an aqueous solution of 4-nitrophenol (5 mL, 0.027 mmol), chloroform solutions of gold nanoparticle catalysts were added (1.8 mL, 0.116 μmol of gold atoms). To this reaction mixture, aqueous sodium borohydride (4 mL, 102.4 mg, 2.707 mmol) was added and the biphasic mixture was vigorously stirred at room temperature under ambient conditions. Aliquots (0.1 mL) were taken from the reaction mixture at various time periods, diluted with water (2.9 mL) and analysed by UV-vis spectroscopy to probe the progress of the reaction. Consistent with the literature,27–30 the UV-vis spectra of the aqueous layer showed that, over time, the absorbance at 400 nm due to the 4-nitrophenolate ion reduced substantially with the concomitant growth of a new peak at 297 nm due to the formation of 4-aminophenol (Fig. 5a).
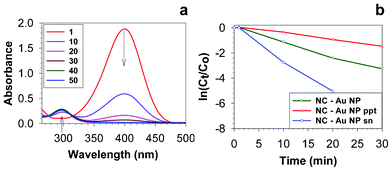 | ||
| Fig. 5 (a) Time-dependent (in min) UV-vis spectra of the 4-nitrophenol reduction reaction catalysed by NC–Au NP. (b) The time-evolution of ln(Ct/Co) of 4-nitrophenol in its reduction catalysed by NC–Au NP, NC–Au NP ppt and NC–Au NP sn. | ||
With NC–Au NP as catalysts, the reduction was complete in ∼40 min (Fig. 5a). Biphasic reactions carried out in the absence of gold catalyst did not show any reduction in the absorbance of 4-nitrophenolate ion during the entire course of the experiment. The as prepared NC–Au NP used in this experiment contains both nanoparticle aggregates and individual nanoparticles. To compare the catalytic abilities of the nanoparticle aggregates and individual nanoparticles, similar catalysis experiments (with identical gold atom concentrations) were carried out with redispersed precipitate (NC–Au NP ppt) and supernatant (NC–Au NP sn), which were complete in 60 and 20 min, respectively. The longer time taken by the nanoparticle aggregates to complete the reduction is not entirely surprising, as other encapsulated metal nanoparticles have also taken up to 60 min to completely reduce 4-nitrophenol.29,31 The plot of ln(Ct/Co) vs. time (Fig. 5b) showed a linear relationship, indicating that these reactions followed first-order kinetics. The ratio of Ct/Co was obtained by the ratio of absorbance at 400 nm at various time periods. Consistent with literature,30 we observed a short induction period at the very initial stages of the reaction after which it followed first order kinetics (Fig. 5b). The rate constants obtained from the linear slope of this plot (in between 1–30 min in Fig. 5b) were 1.9 × 10−3 s−1 for NC–Au NP, 0.8 × 10−3 s−1 for NC–Au NP (redispersed precipitate) and 4.3 × 10−3 s−1 for NC–Au NP sn (supernatant) samples. Diffusion could play a major role in the catalysis mediated by nanocapsule encapsulated nanoparticle aggregates,29,32 and we are currently investigating the permeation characteristics of these resorcinarene nanocapsules in detail.
In conclusion, we have shown that the in situ synthesis and templating of metal nanoparticles by RTATT nanocapsules can be carried out with high structural fidelity and nanoparticle loading, along with the potential to control the resulting nanostructures. Though we have demonstrated their utility in the fabrication of Au nanostructures, these nanocapsules can be readily used for fabricating other metallic nanostructures. Currently, we are working on the synthesis of metal nanostructures using other morphologically distinct resorcinarene polymer templates.14 We have investigated the utility of nanocapsule templated nanoparticle aggregates in the reduction of 4-nitrophenol. Controlled formation of metallic nanostructures with tunable optical properties is expected to play a crucial role in SERS based applications.10,23
The authors gratefully acknowledge the multidisciplinary seed grant by the Office of Research of Old Dominion University (ODU) for financial support. The authors thank Professors Jennifer Poutsma and Kenneth Brown, both from ODU, for valuable inputs in the preparation of this manuscript. Thanks are also due to Professor Anna H. Jeng, ODU, for access to the Malvern Zetasizer. We also thank the Reviewers of this manuscript for valuable suggestions.
References
- M. R. Jones, K. D. Osberg, R. J. Macfarlane, M. R. Langille and C. A. Mirkin, Chem. Rev., 2011, 111, 3736–3827 CrossRef CAS.
- Z. H. Nie, A. Petukhova and E. Kumacheva, Nat. Nanotechnol., 2010, 5, 15–25 CrossRef CAS.
- M. Grzelczak, J. Vermant, E. M. Furst and L. M. Liz-Marzan, ACS Nano, 2010, 4, 3591–3605 CrossRef CAS.
- S. Kinge, M. Crego-Calama and D. N. Reinhoudt, ChemPhysChem, 2008, 9, 20–42 CrossRef CAS.
- R. Shenhar, T. B. Norsten and V. M. Rotello, Adv. Mater., 2005, 17, 657–669 CrossRef CAS.
- A. C. Balazs, T. Emrick and T. P. Russell, Science, 2006, 314, 1107–1110 CrossRef CAS.
- L. L. del Mercato, P. Rivera-Gil, A. Z. Abbasi, M. Ochs, C. Ganas, I. Zins, C. Sonnichsen and W. J. Parak, Nanoscale, 2010, 2, 458–467 RSC.
- L. J. De Cock, S. De Koker, B. G. De Geest, J. Grooten, C. Vervaet, J. P. Remon, G. B. Sukhorukov and M. N. Antipina, Angew. Chem., Int. Ed., 2010, 49, 6954–6973 CrossRef CAS.
- K. Zhang, H. Fang, Z. Li, J. Ma, S. V. Hohlbauch, J. S. A. Taylor and K. L. Wooley, Soft Matter, 2009, 5, 3585–3589 RSC.
- M. Sanles-Sobrido, W. Exner, L. Rodríguez-Lorenzo, B. Rodríguez-González, M. A. Correa-Duarte, R. A. Álvarez-Puebla and L. M. Liz-Marzán, J. Am. Chem. Soc., 2009, 131, 2699–2705 CrossRef CAS.
- G. L. Li, L. Q. Xu, K. G. Neoh and E. T. Kang, Macromolecules, 2011, 44, 2365–2370 CrossRef CAS.
- S. N. Shmakov and E. Pinkhassik, Chem. Commun., 2010, 46, 7346–7348 RSC.
- H. Li, C.-S. Ha and I. Kim, Macromol. Rapid Commun., 2009, 30, 188–193 CrossRef CAS.
- R. Balasubramanian, Z. M. Kalaitzis and W. Cao, J. Mater. Chem., 2010, 20, 6539–6543 RSC.
- R. Balasubramanian and Z. M. Kalaitzis, ACS Symp. Ser., 2011, 1070, 263–276 CrossRef CAS.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802 RSC.
- D. Kim, E. Kim, J. Lee, S. Hong, W. Sung, N. Lim, C. G. Park and K. Kim, J. Am. Chem. Soc., 2010, 132, 9908–9919 CrossRef CAS.
- S. J. Oldenburg, R. D. Averitt, S. L. Westcott and N. J. Halas, Chem. Phys. Lett., 1998, 288, 243–247 CrossRef CAS.
- R. Bardhan, N. K. Grady, T. Ali and N. J. Halas, ACS Nano, 2010, 4, 6169–6179 CrossRef CAS.
- W. Shi, Y. Sahoo, M. T. Swihart and P. N. Prasad, Langmuir, 2005, 21, 1610–1617 CrossRef CAS.
- C. Graf and A. van Blaaderen, Langmuir, 2001, 18, 524–534 CrossRef.
- A. M. Schwartzberg, T. Y. Olson, C. E. Talley and J. Z. Zhang, J. Phys. Chem. B, 2006, 110, 19935–19944 CrossRef CAS.
- M. Yang, R. N. Alvarez-Puebla, H.-S. Kim, P. Aldeanueva-Potel, L. M. Liz-Marzán and N. A. Kotov, Nano Lett., 2010, 10, 4013–4019 CrossRef CAS.
- H. M. Chen, R.-S. Liu, K. Asakura, J.-F. Lee, L.-Y. Jang and S.-F. Hu, J. Phys. Chem. B, 2006, 110, 19162–19167 CrossRef CAS.
- M. J. Hostetler, J. E. Wingate, C.-J. Zhong, J. E. Harris, R. W. Vachet, M. R. Clark, J. D. Londono, S. J. Green, J. J. Stokes, G. D. Wignall, G. L. Glish, M. D. Porter, N. D. Evans and R. W. Murray, Langmuir, 1998, 14, 17–30 CrossRef CAS.
- C. Y. Song, G. P. Zhao, P. J. Zhang and N. L. Rosi, J. Am. Chem. Soc., 2010, 132, 14033–14035 CrossRef CAS.
- S. Saha, A. Pal, S. Kundu, S. Basu and T. Pal, Langmuir, 2009, 26, 2885–2893 CrossRef.
- N. Hu, J.-Y. Yin, Q. Tang and Y. Chen, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3826–3834 CrossRef CAS.
- J. Lee, J. C. Park and H. Song, Adv. Mater., 2008, 20, 1523–1528 CrossRef CAS.
- S. Wunder, F. Polzer, Y. Lu, Y. Mei and M. Ballauff, J. Phys. Chem. C, 2010, 114, 8814–8820 CAS.
- A. A. Antipov, G. B. Sukhorukov, Y. A. Fedutik, J. Hartmann, M. Giersig and H. Möhwald, Langmuir, 2002, 18, 6687–6693 CrossRef CAS.
- Y. Lvov, A. A. Antipov, A. Mamedov, H. Möhwald and G. B. Sukhorukov, Nano Lett., 2001, 1, 125–128 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |