Salt/pH dual-responsive supramolecular brush copolymer micelles with molecular recognition of nucleobases for drug delivery†
Dali
Wang
a,
Xiuying
Huan
a,
Lijuan
Zhu
a,
Jinyao
Liu
a,
Feng
Qiu
a,
Deyue
Yan
a and
Xinyuan
Zhu
*ab
aSchool of Chemistry and Chemical Engineering, State Key Laboratory of Metal Matrix Composites, Shanghai Jiao Tong University, 800 Dongchuan Road, Shanghai 200240, People's Republic of China
bInstrumental Analysis Center, Shanghai Jiao Tong University, 800 Dongchuan Road, Shanghai 200240, People's Republic of China. E-mail: xyzhu@sjtu.edu.cn; Tel: +86-21-34205699; Fax: +86-21-34205722
First published on 29th October 2012
Abstract
A new type of salt/pH dual-responsive micelles based on supramolecular amphiphilic brush copolymers, poly(2-hydroxyethyl metharylate)-g-(poly(ε-caprolactone)-adenine:uracil-poly(ethylene glycol)) (PHEMA-g-(PCL-A:U-PEG)) was developed for the anticancer drug delivery owing to the fact that the tumor tissues show low pH and high salt concentration. The supramolecular structure of brush copolymer was confirmed by variable-temperature 1H NMR and Fourier transform infrared spectroscopy (FTIR). Doxorubicin (DOX) as a model anticancer drug was efficiently loaded into the supramolecular micelles (up to 70%) due to the compact structure of brush polymer. The cumulative release profile of the DOX-loaded micelles showed a low level of drug release (about 20 wt% in 25 h) at pH 7.4 with a low salt concentration, and was significantly accelerated at a lower pH (5.0) and a high salt concentration (over 70 wt% in 6 h), exhibiting an salt/pH dual-responsive controlled drug release capability. Methyl tetrazolium (MTT) assay showed that DOX-loaded micelles had high anticancer efficacy against Hela cancer cells and blank micelles had a very low cytotoxicity. These supramolecular brush copolymer micelles possess many favorable traits, such as low cytotoxicity and excellent biodegradability, adequate drug loading capacity, and rapid drug release in response to the intracellular level of pH and salt concentration, which endow them as a promise candidate for delivering anticancer drugs.
Introduction
As one of the most promising nanocarrier systems in disease therapy, stimuli-responsive polymeric micelles have attracted significant attention over the past two decades.1–6 To realize an efficient delivery of drugs, there are two basic requirements for the design of “smart” polymeric drug carriers.7,8 Firstly, the polymer carrier should respond rapidly to intracellular stimulus and consequently release the drugs in the pathologic area. Conditions within the tumor microenvironment differ considerably from those in normal tissue. For example, the tumor extracellular environment is more acidic (pH 6.5) than blood and normal tissues (pH 7.4), and the pH values of endosome and lysosome are even lower at 5.0–5.5.9,10 To date, various pH-responsive polymer micelles based on acid-sensitive groups have been developed for pH-triggered drug delivery.11–17 Most of these delivery systems are responsive to a single pH condition, impairing the drug delivery efficacy and making them inefficient in killing drug-resistant cells. In fact, sodium and chlorine concentrations are significantly higher in tumorous cells than in any of the other cell populations (up to 451 ± 6 (S.E.)).18,19 High intracellular sodium and chlorine concentrations are associated with oncogenesis. These factors make salt an ideal candidate as a therapeutic target for the development of salt-responsive drug delivery systems. Providing that the polymeric carriers respond not only to acidic pH, but also to high salt concentration, rapid drug release in tumor sites will be achieved. Secondly, a well-designed polymeric nanocarrier should have a high loading efficiency. Low drug loading content causes high drug cost and low therapeutic efficacy. It has been reported that the architecture of polymers may have a great effect on the size, morphology and stability of micelles, as well as other aspects of the drug delivery, including drug loading efficiency and release kinetics, even in vivo circulation and distribution.20,21 Therefore, it is of great importance to optimize polymer architecture to fabricate novel drug delivery systems. Considering these two requirements, design and preparation of polymer carriers with salt/pH dual-responsive ability and high drug encapsulation become very attractive.As a newly emerging smart material, supramolecular polymers based on noncovalent interactions have been found to be more sensitive to external stimuli, which offers a new route for the design of drug delivery systems with rapid response abilities.22–24 In particular, complementary hydrogen bonds from nucleobases, which are moderately strong, highly directional and sensitive to not only acidic pH, but also salt, show great potential in biomedical applications.25–28 In our previous work, we tried to utilize base pairs to fabricate supramolecular linear copolymer micelles for drug delivery.29 It has been proved that complementary multiple hydrogen bonding has potential as a “smart” linker. However, the linear supramolecular copolymer micelles suffered from thermodynamic instability, due to their relatively high critical micelle concentration (CMC), and drug loading and efficiency were very low, which limited therapeutic efficacy against solid tumors and gave difficulty in clinical applications. In the present work, we designed salt/pH dual-responsive supramolecular brush polymer micelles based on molecular recognition of nucleobases for intracellular drug delivery. Brush polymers were chosen, because they have compact structure and a consistent cylindrical shape, which are good candidate properties for encapsulating drug molecules efficiently.30,31 Compared with supramolecular linear copolymer micelles, supramolecular brush copolymer micelles exhibited the following advantages: (1) due to their compact architecture, they exhibited higher drug loading content and efficiency, thereby providing a sufficient concentration of doxorubicin (DOX) in the tumor cells; (2) they show lower CMC, which is beneficial for the stability of micelles, and (3) supramolecular brush copolymers could self-assemble into smaller micelles, which could be rapidly localized intracellularly and subsequently provoked by the acidic pH and high salt concentration to release the drug, leading to higher anticancer efficacy with reduced side effects. To our knowledge, this is the first attempt to prepare supramolecular brush copolymer micelles based on molecular recognition of nucleobases for drug delivery. The in vitro study of the drug-loaded micelles indicated the great potential of supramolecular brush copolymer micelles as a smart drug carrier for triggered intracellular drug delivery.
Experimental section
Materials
2-Hydroxyethyl methacrylate (HEMA, 97%, Aldrich) was purified according to the literature.32 Copper(I) bromide (CuBr) was purified by stirring in acetic acid and washing with ethanol, and then dried under vacuum. ε-Caprolactone (ε-CL, 99%, Alfa Aesar) was dried over calcium hydride (CaH2) for 24 h and then purified by distillation under reduced pressure prior to use. Ethyl 2-bromoisobutyrate (EBiB, 98%, Acros), 2,2′-bipyridine (bpy, 99%, Aldrich), Tin(II) octoate (Sn(Oct)2), uracil (U, 99%), adenine (A, 99%) and 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) were all purchased from Sigma and used as received. 1-Ethyl-3-(3-dimethyllaminopropyl)-carbodiimide hydrochloride (EDC) and 4-dimethylamino-pyridine (DMAP) were purchased from Aladdin. N,N-Dimethylformamide (DMF) was dried over CaH2 for 48 h and then distilled before use. Toluene was dried by refluxing with fresh sodium-benzophenone complex under nitrogen gas and distilled just before use. Monomethoxy poly(ethylene glycol) (PEG) with Mn = 2000 g mol−1 was purchased from Fluka and dried by azeotropic distillation in the presence of dry toluene. Doxorubicin hydrochloride (DOX·HCl) was purchased from Beijing Huafeng United Technology Corporation and used as received. Clear polystyrene tissue culture treated 12-well and 96-well plates were obtained from Corning Costar. All other reagents and solvents were purchased from the domestic suppliers and used as received unless otherwise mentioned. 3-(9-Adeninyl)propionic acid was prepared according to the previous literature.33,34Characterization
All nuclear magnetic resonance (NMR) spectra were recorded on a Bruker AVANCEIII 400 spectrometer with deuterated chloroform (CDCl3) and dimethyl sulfoxide-d6 (DMSO-d6) as solvents. The number-average molecular weight (Mn), weight-average molecular weight (Mw) and polydispersity index (PDI) were determined by gel permeation chromatography (GPC). GPC was performed on a Perkin-Elmer series 200 system (10 μm PL gel 300 × 7.5 mm mixed-B and mixed-C column, linear polystyrene calibration) equipped with a refractive index (RI) detector. DMF containing 0.01 mol L−1 lithium bromide was used as the mobile phase at a flow rate of 1 mL min−1 at 70 °C. Fourier transform infrared (FTIR) spectra were recorded on a Paragon 1000 instrument by the KBr sample holder method. Dynamic light scattering (DLS) measurements were performed with a Malvern Zetasizer Nano S apparatus equipped with a 4.0 mW laser operating at λ = 633 nm. All samples were measured at a scattering angle of 173°. The data presented are the mean of three tests. Transmission electron microscopy (TEM) studies were performed with a JEOL JEM-100CX-II instrument at a voltage of 200 kV. Samples were prepared by drop-casting micelle solutions onto carbon-coated copper grids and then air-drying at room temperature before measurement.Synthesis of poly(2-hydroxyethyl metharylate) (PHEMA)
In a typical experiment, a dry Schlenk flask with a magnetic stirrer was charged with HEMA monomer (57 mmol, 6.88 mL), the initiator EBiB (0.38 mmol, 56 μL), bpy (0.76 mmol, 119.3 mg) and 10 mL of methanol (HPLC grade). The flask was then degassed with three freeze-thaw cycles, followed by adding CuBr (0.38 mmol, 54.8 mg) under the protection of a nitrogen atmosphere. The polymerization was conducted at 40 °C and stopped by exposing the solution to air at about 40% monomer conversion. The resulting solution was passed through a neutral alumina column to remove the copper catalysts. The polymer was precipitated twice in diethyl ether and dried in vacuo at room temperature for 48 h. Mn,GPC = 1.89 × 104 g mol−1, Mw/Mn = 1.30.1H NMR (400 MHz, DMSO-d6, 298 K) δ (ppm): 4.8 (1H, s, –CH2OH); 3.9 (2H, s, –O–CH2–CH2–); 3.6 (2H, s, –O–CH2–CH2–); 2.0–1.7 (2H, d, –CH2–C(CH3)–); 1.0–0.7 (3H, d, –CH2–C(CH3)–).
Synthesis of poly(2-hydroxyethyl metharylate)-g-(poly(ε-caprolactone) (PHEMA-g-PCL)
A typical polymerization procedure was as follows. In a fire-dried glass flask, PHEMA (0.22 g, 1.69 mmol –OH) was dissolved in 1.5 mL of freshly distilled anhydrous DMF. Then ε-CL (4.82 g, 42.28 mmol) and Sn(Oct)2 (11.0 mg, 0.027 mmol) were added to the solution, and the flask was immersed in an oil bath at 115 °C for 24 h. After cooled to room temperature, the resulting polymer was dissolved in dichloromethane and precipitated twice from methanol. The purified copolymer was dried under vacuum to a constant weight. Mn,GPC = 15.3 × 104 g mol−1, Mw/Mn =1.3.
1H NMR (400 MHz, CDCl3, 298 K) δ (ppm): 3.9–4.2 (polycaprolactone, –COOCH2–), 3.6 (polycaprolactone, –CH2CH2–OH), 2.2–2.4 (polycaprolactone, –CO![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2CH2–), 1.5–1.8 (polycaprolactone, –COCH2CH2–, –COO–CH2CH2–), 1.2–1.4 (polycaprolactone, –COCH2CH2CH2–).
CH2CH2–), 1.5–1.8 (polycaprolactone, –COCH2CH2–, –COO–CH2CH2–), 1.2–1.4 (polycaprolactone, –COCH2CH2CH2–).
Synthesis of poly(2-hydroxyethyl metharylate)-g-(poly(ε-caprolactone)-adenine) (PHEMA-g-(PCL-A))
PHEMA-g-(PCL-A) was prepared by coupling 3-(9-adeninyl)propionic acid with PHEMA-g-PCL. In a typical procedure, PHEMA-g-PCL (1.0 g, 0.75 mmol OH), 3-(9-adeninyl)propionic acid (0.23 g, 1.10 mmol), EDC (0.42 g, 2.20 mmol), and DMAP (0.136 g, 1.10 mmol) were dissolved in DMF (60 mL) and stirred at 45 °C for 24 h. DMF was evaporated under reduced pressure, and the residue was precipitated into cold methanol three times. The product was dried under vacuum to a constant weight. Mn,GPC = 17.0 × 104 g mol−1, Mw/Mn =1.6.
1H NMR (400 MHz, CDCl3, 298 K) δ (ppm): 8.3 (1H, –NCHN–), 7.9 (1H, NCHN–), 4.5 (2H, –NCH2CH2–), 3.9–4.2 (polycaprolactone, –COOCH2–), 2.9 (2H, –NCH2CH2–), 2.2–2.4 (polycaprolactone, –COCH2CH2–), 1.5–1.8 (polycaprolactone, –COCH2CH2–, –COO–CH2CH2–), 1.2–1.4 (polycaprolactone, –COCH2CH2CH2–).
Synthesis of uracil-terminated PEG (PEG-U)
PEG-U was prepared via a two-step synthesis. First, PEG–sulfanilic acid ester was prepared by reacting PEG with excess p-toluenesulfonyl chloride (TsCl) in the presence of sodium hydroxide (NaOH). Then PEG–sulfanilic acid ester (1.60 g, 0.8 mmol), uracil (0.18 g, 1.6 mmol) and potassium carbonate (K2CO3) (0.22 g, 1.6 mmol) were dissolved in 30 mL of freshly distilled anhydrous DMF in a round-bottomed flask and reacted at 80 °C for 8 h. The resultant mixture was poured into water and extracted with CH2Cl2. After drying over anhydrous magnesium sulfate (MgSO4), most of the solvent was removed by evaporation. The residue was precipitated into cold diethyl ether and dried under vacuum to a constant weight. 1H NMR (400 MHz, CDCl3, 298 K) δ (ppm): 8.4 (1H, –CONHCO–), 7.4 (1H, –NCHCH–), 5.6 (1H, –CHCH–CO–), 3.9 (2H, –CH2CH2N–), 3.5–3.7 (PEG, 2H per repeating unit, –OCH2CH2–, 2H per repeating unit, –O–CH2CH2–), 3.4 (3H, CH3–OCH2–). 13C NMR (100 MHz, CDCl3, 298 K) δ: 47.0 (–CH2CH2N–), 58.0 (CH3O–), 67.8 (–CH2CH2N–), 69.7 (–CH2CH2O–), 71.2 (CH3OCH2–), 100.2 (–CHCHCO–), 146.3 (–CHCHCO–), 150.9 (–NCONH–), 163.8 (–CHCONH–).Preparation of PHEMA-g-(PCL-A:U-PEG) micelles and determination of their CMC
Micelles were prepared by a dialysis method. Typically, PHEMA-g-(PCL-A:U-PEG) copolymer (20 mg, equivalent molar PCL-A and PEG-U) were dissolved in THF (2 mL) and stirred for 4 h at room temperature. After that, the polymer solution was added dropwise into 10 mL of deionized water under stirring with a magnetic bar. Then the micelle solution was dialyzed against deionized water for 24 h (molecular weight cut off (MWCO) = 2000 Da) to remove the organic solvent, during which the water was renewed every 4 h.The CMC was determined using 1,6-diphenyl-1,3,5-hexatriene (DPH) as a UV probe by monitoring the absorbance at 313 nm. The concentration of supramolecular copolymer was varied from 5.0 × 10−5 to 0.5 mg mL−1 and the DPH concentration was fixed at 5.0 × 10−6 mol L−1. The absorbance spectra of all solutions were recorded using a Perkin-Elmer Lambda 20/2.0 UV/Vis spectrometer.
Preparation of DOX-loaded PHEMA-g-(PCL-A:U-PEG) micelles
Briefly, DOX·HCl and an equal molar amount of triethylamine (TEA) were dissolved in DMSO and added to a THF solution of PHEMA-g-(PCL-A:U-PEG) at a theoretical drug loading content of 10 wt%. Then the mixture was added slowly to 5 mL phosphate buffered saline (PBS, 50 mM, pH 7.4). After being stirred for an additional 4 h, the solution was dialyzed against deionized water for 24 h (MWCO = 2000 Da), during which the water was renewed every 4 h. For determination of drug loading content, the DOX-loaded micelle solution was lyophilized and then dissolved in DMF. The UV absorbance at 485 nm was measured to determine the DOX concentration.Drug loading content (DLC) and drug loading efficiency (DLE) were calculated according to the following formulae:
DLC (wt%) = (weight of loaded drug/weight of polymer) × 100%
DLE (%) = (weight of loaded drug/weight of drug in feed) × 100%
In vitro release study
A total of 6 mL of DOX-loaded PHEMA-g-(PCL-A:U-PEG) micelles was transferred to a dialysis bag with a MWCO of 2000 Da. It was immersed in 50 mL of deionized water, phosphate buffer (pH 7.4, Na+ 0.12 M or 0.56 M) and acetate buffer (pH 5.0, Na+ 0.12 M or 0.56 M) solutions with gentle shaking (100 rpm) at 37 °C in a laboratory shaker. At predetermined time intervals, 2.0 mL solution outside the dialysis bag was extracted and replaced by an equal volume of fresh media to retain the sink condition. The amount of released DOX was analyzed using fluorescence (QM/TM/IM Steady-State & Time-Resolved Fluorescence Spectrofluorometer) measurement (excitation at 480 nm). Each experiment was performed in triplicate and the presented results are the average of this data.Cell culture
NIH/3T3 normal cells (a mouse embryonic fibroblast cell line) and Hela cancer cells (a human uterine cervix carcinoma cell line) were cultured in DMEM (Dulbecco's modified Eagle's medium) supplied with 10% FBS (fetal bovine serum) and antibiotics (50 units mL−1 penicillin and 50 units mL−1 streptomycin) at 37 °C in a humidified atmosphere containing 5% CO2.Cytotoxicity measurements of micelles
The relative cytotoxicity of PHEMA-g-(PCL-A:U-PEG) micelles against NIH/3T3 cells was estimated by MTT viability assay. In the MTT assay, NIH/3T3 cells were seeded into 96-well plates with a density of 1 × 104 cells per well in 200 μL of medium. After 24 h of incubation, the culture medium was removed and replaced with 200 μL of a medium containing serial dilutions of micelles. The cells were grown for another 24 h. Then, 20 μL of 5 mg mL−1 MTT assay stock solution in PBS was added to each well. After incubating the cells for 4 h, the medium containing unreacted dye was removed carefully. The obtained blue formazan crystals were dissolved in 200 μL per well of DMSO and the absorbance was measured in a BioTek® SynergyH4 at a wavelength of 490 nm.Cytotoxicity measurements of micelles
The cellular uptake experiments were performed using flow cytometry and confocal laser scanning microscopy (CLSM). For flow cytometry, Hela cells were seeded in 6-well plates at 6 × 105 cells per well in 1 mL complete DMEM and cultured for 24 h. Then, the DOX-loaded micelles dissolved in DMEM culture medium, at a final DOX concentration of 9 μg mL−1, were added to different wells and the cells were incubated at 37 °C for 5, 15, 30, 60, and 180 min. Thereafter, samples were prepared for flow cytometry analysis by removing the cell growth media, rinsing with PBS, and treating with trypsin. Data for 1.0 × 104 gated events were collected and analysis was performed by means of a BD FACSCalibur flow cytometer and CELLQuest software.For the CLSM studies, Hela cells were seeded in 6-well plates at 1 × 105 cells per well in 1 mL complete DMEM and cultured for 24 h, followed by removing culture medium and adding DOX-loaded micelles (1 mL DMEM medium) at a final DOX concentration of 9 μg mL−1. The cells were incubated at 37 °C for the predetermined intervals. Subsequently, the cells were washed with PBS and fixed with 4% paraformaldehyde for 30 min at room temperature, and the slides were rinsed with PBS for three times. Finally, the slides were mounted and observed with a LSM510 META.
Activity analyses
The cytotoxicity of DOX-loaded micelles and free DOX against Hela cells was evaluated in vitro by MTT assay. Hela cells were seeded into 96-well plates with a density of 8 × 103 cells per well in 200 μL of medium. After 24 h of incubation, the culture medium was removed and replaced with 200 μL of a medium containing serial dilutions of DOX-loaded micelles. The cells were grown for another 48 h. Then, 20 μL of 5 mg mL−1 MTT assays stock solution in PBS was added to each well. After incubating the cells for 4 h, the medium containing unreacted dye was removed carefully. The obtained blue formazan crystals were dissolved in 200 μL per well DMSO and the absorbance was measured in a BioTek® SynergyH4 at a wavelength of 490 nm.Results and discussion
Synthesis and characterization of PHEMA-g-(PCL-A) and PEG-U
The synthesis route of PHEMA-g-(PCL-A) and PEG-U is schematically illustrated in Scheme 1. First, atom transfer radical polymerization (ATRP) was used to directly prepare PHEMA with a controlled molecular weight and low polydispersity. The polymerization was performed in methanol using a CuBr/bpy catalyst system and EBiB as an initiator. The initial ratio of monomer to initiator was 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, and polymerization was stopped when monomer conversion was about 40% (degree of polymerization (DP) 60). 1H NMR spectrum of PHEMA in Fig. 1A confirmed the successful synthesis. The molecular weight and molecular weight distribution of the resulting PHEMA were determined by GPC using DMF as the eluent (Fig. 2). The apparent molecular weight obtained by GPC polystyrene (PS) standards (Mn = 18 900 g mol−1) was higher than the theoretical molecular weight (Mn,theory = 8 000 g mol−1) calculated from the HEMA conversion (Mn,theory = conversion × MWHEMA × [HEMA]0/[EBiB]0) due to the differences in hydrodynamic volumes of PHEMA and PS standards.35,36
:1, and polymerization was stopped when monomer conversion was about 40% (degree of polymerization (DP) 60). 1H NMR spectrum of PHEMA in Fig. 1A confirmed the successful synthesis. The molecular weight and molecular weight distribution of the resulting PHEMA were determined by GPC using DMF as the eluent (Fig. 2). The apparent molecular weight obtained by GPC polystyrene (PS) standards (Mn = 18 900 g mol−1) was higher than the theoretical molecular weight (Mn,theory = 8 000 g mol−1) calculated from the HEMA conversion (Mn,theory = conversion × MWHEMA × [HEMA]0/[EBiB]0) due to the differences in hydrodynamic volumes of PHEMA and PS standards.35,36
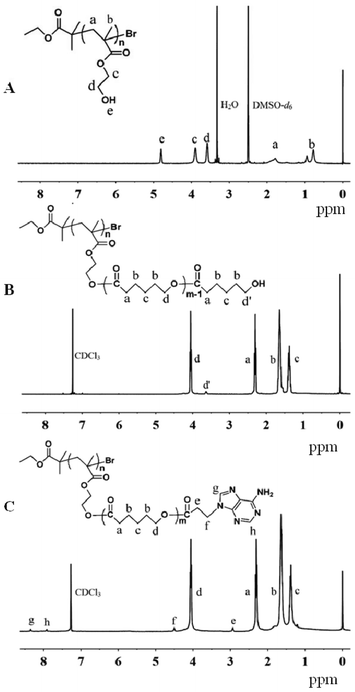 | ||
| Fig. 1 1H NMR spectra of (A) PHEMA, (B) PHEMA-g-PCL and (C) PHEMA-g-(PCL-A). | ||
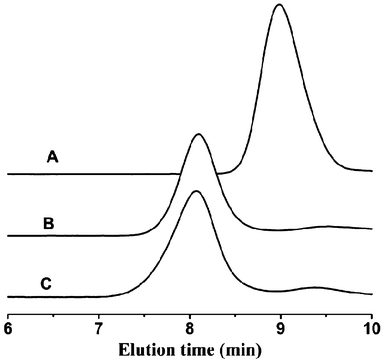 | ||
| Fig. 2 GPC curves of (A) PHEMA, (B) PHEMA-g-PCL and (C) PHEMA-g-(PCL-A). | ||
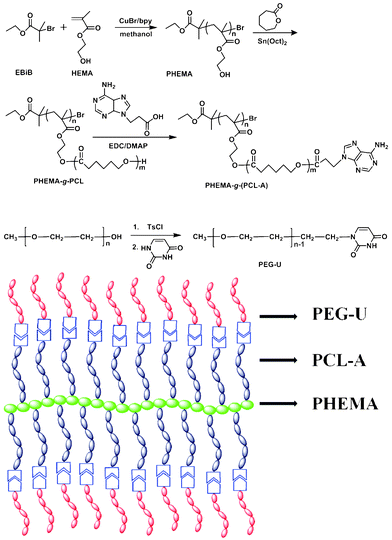 | ||
| Scheme 1 Synthesis of PHEMA-g-(PCL-A) brush macromolecule and PEG-U by ATRP, ROP and coupling reaction and schematic structure of supramolecular brush copolymer PHEMA-g-(PCL-A:U-PEG). | ||
In the next step, PHEMA-g-(PCL-A) was prepared by a combination of ring-opening polymerization (ROP) and coupling reaction. PHEMA-g-PCL was first synthesized by ROP using PHEMA as the macroinitiator for ε-CL polymerization in dried DMF under the catalysis of Sn(Oct)2 at 115 °C for 24 h. The coupling reaction of 3-(9-adeninyl)propionic acid to PHEMA-g-PCL was performed at 45 °C in the presence of EDC and DMAP. In the coupling reaction, it was not necessary to protect the primary amino groups in the adenine derivate, because they are reasonably deactivated due to the resonance effect originating from the two conjugated rings.34 The structures of PHEMA-g-PCL and PHEMA-g-(PCL-A) were confirmed by 1H NMR (Fig. 1B and 1C). According to the relative integral values of the methylene signal on the PCL polymer backbone (–CH2OCO–, 3.9–4.2 ppm) and that connected with adenine (–N–CH2CH2–, 4.5 ppm) from the 1H NMR spectra, the coupling efficiency of PHEMA-g-(PCL-A) is calculated to be 72%. The molecular weights of PHEMA-g-PCL and PHEMA-g-(PCL-A) were calculated by 1H NMR spectrum and measured by GPC, respectively. The data are summarized in Table 1. The representative GPC profiles of PHEMA-g-PCL and PHEMA-g-(PCL-A) are shown in Fig. 2. The GPC analysis of these reveals a relatively unimodal and symmetric elution peak. A clear shift toward higher molecular weight is observed by comparing the GPC curves of PHEMA, PHEMA-g-PCL and PHEMA-g-(PCL-A). This indicates that the corresponding coupling reaction is successful.
| Polymers | M n a (g mol−1;×104) | M n b (g mol−1; ×104) | M w b (g mol−1; ×104) | M w/Mnb |
|---|---|---|---|---|
| a Determined by 1H NMR. b Determined by GPC using DMF as the eluent. c Theoretical molecular weight determined from monomer conversions. | ||||
| PHEMA | 0.80c | 1.89 | 2.46 | 1.3 |
| PHEMA-g-PCL | 14.6 | 15.3 | 20.8 | 1.3 |
| PHEMA-g-(PCL-A) | 15.5 | 17.0 | 28.1 | 1.6 |
PEG-U was prepared in a two-step reaction. First, PEG–sulfanilic acid ester was prepared by the coupling reaction of PEG and TsCl. Then PEG-U was obtained by the grafting reaction of PEG–sulfanilic acid ester and an excess of uracil. A detailed characterization of PEG-U can be found in the experimental section.
Hydrogen bonding interactions between PHEMA-g-(PCL-A) and PEG-U
The proposed molecular model of supramolecular brush copolymer PHEMA-g-(PCL-A:U-PEG) is illustrated in Scheme 1. The formation of complementary multiple hydrogen bonds between PHEMA-g-(PCL-A) and PEG-U was analyzed by variable-temperature 1H NMR spectroscopy for the blend of two polymers with equivalent molar quantities of A and U in 1,1,2,2-tetrachloroethane-d2. The temperature dependence of the NH proton chemical shift of the PHEMA-g-(PCL-A) and PEG-U complex at 3.5 wt% in 1,1,2,2-tetrachloroethane-d2 is shown in Fig. 3. The chemical shift of the NH resonance moves upfield systematically from 8.4 to 7.9 ppm when the temperature is raised from 25 to 100 °C. The gradual decrease in the NH resonance with the increase of temperature can be attributed to the dissociation of the complementary hydrogen bonds.37 However, as soon as the sample is cooled from 100 to 25 °C, the NH resonance returns to its original position at 8.4 ppm. This suggests that the complementary hydrogen bonds generated between PHEMA-g-(PCL-A) and PEG-U, and the supramolecular brush copolymers are thermo-reversible in 1,1,2,2-tetrachloroethane-d2.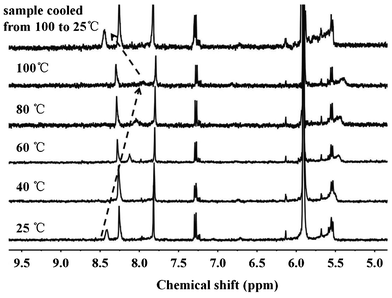 | ||
| Fig. 3 1H NMR NH chemical shift of the PHEMA-g-(PCL-A) and PEG-U complex (an equimolar ratio of A and U) as a function of temperature in 1,1,2,2-tetrachloroethane-d2. Sample was allowed to equilibrate for 5 min at each temperature. | ||
The molecular recognition of nucleobases between PHEMA-g-(PCL-A) and PEG-U was also analyzed by variable-temperature FTIR. The partial IR spectra of PHEMA-g-(PCL-A) and PEG-U complexes with equivalent molar adenine and uracil units in the bulk state are shown in Fig. S1.† As the temperature increases from 25 to 180 °C, the bands in the range of 1750–1710 cm−1, belonging to the CO stretching vibration, shift to a higher frequency, from 1724 to 1735 cm−1. When the sample is cooled from 180 to 25 °C, the CO stretching peak returns to its original position at 1724 cm−1. According to previous reports, the CO stretching bands shift to lower frequencies upon hydrogen bond formation.38 The results of the FTIR spectra suggest the existence of hydrogen bonds between PHEMA-g-(PCL-A) and PEG-U and that these complementary hydrogen bonds dissociate with increasing temperature. It can be concluded that PHEMA-g-(PCL-A) and PEG-U are linked through complementary hydrogen bonds to form a supramolecular brush copolymer.
Preparation and characterization of PHEMA-g-(PCL-A:U-PEG) micelles
Due to its amphihphilic nature, the supramolecular brush copolymer PHEMA-g-(PCL-A:U-PEG) can self-assemble into micelles in aqueous solution (Scheme 2). The formation of supramolecular copolymer micelles was confirmed by UV/Vis technique using DPH as a probe. The intensity of absorbance of DPH versus the copolymer concentration is shown in Fig. 4. The intensity remains almost constant at low polymer concentration and increases sharply when the copolymer concentration reaches a value that results in the transfer of DPH molecules from the water environment to the hydrophobic micellar core. It confirms the formation of copolymer micelles and the CMC value of PHEMA-g-(PCL-A:U-PEG) is determined as 0.014 mg mL−1 in aqueous solution. The CMC value was further determined by DLS.39 As shown in Fig. S2,† the first portion of the curve represents the non-aggregated form of PHEMA-g-(PCL-A:U-PEG) in water, or at least smaller aggregates than those observed in the second portion of the curve.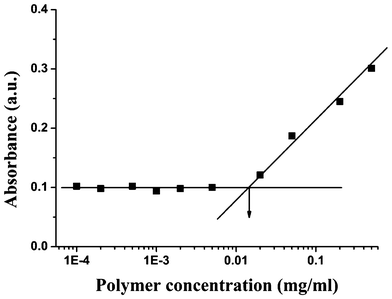 | ||
| Fig. 4 CMC of supramolecular brush copolymer PHEMA-g-(PCL-A:U-PEG). | ||
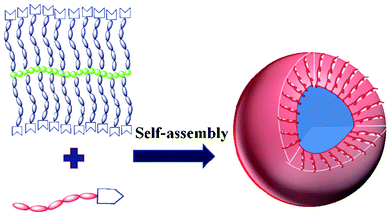 | ||
| Scheme 2 Schematic representation of self-assembly of PHEMA-g-(PCL-A:U-PEG) in water and the core/shell aggregating structure of resulting micelle. | ||
To further evaluate the properties of supramolecular brush copolymer micelles, both TEM and DLS measurements were performed. The morphology of the supramolecular brush copolymer micelles was visualized by TEM. Fig. 5A displays approximate spherical morphology and the average diameter is about 60 nm. The size distribution of the micelles was also determined by DLS, and Fig. 6A gives a monomodal size distribution with a Z-average diameter of 108 nm with a PDI of 0.14. It is worth noting that the diameter of micelles observed by DLS is larger than that determined by TEM measurement. Such a difference is reasonable, as the diameter of the micelles determined by DLS reflects the hydrodynamic diameter of the micelles, whereas the diameter visualized by TEM gives that of dried micelles.
 | ||
| Fig. 5 TEM images of (A) PHEMA-g-(PCL-A:U-PEG) micelles and (B) DOX-loaded PHEMA-g-(PCL-A:U-PEG) micelles. | ||
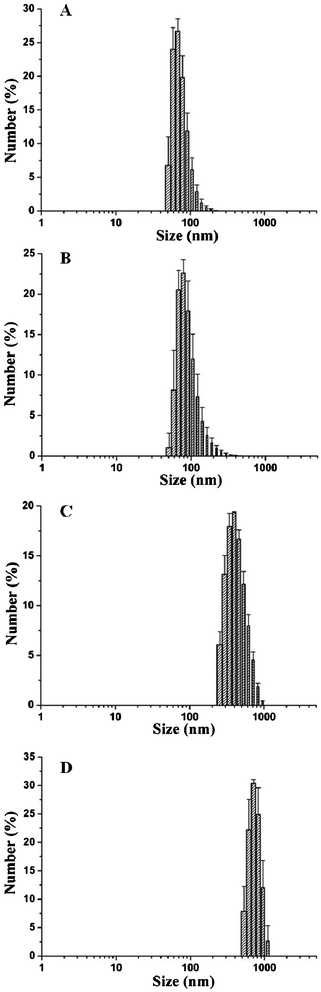 | ||
| Fig. 6 The size distributions of micelles after being exposed to various stimuli: (A) water; (B) pH 7.4, 0.12 M Na+; (C) pH 7.4, 0.56 M Na+, (D) pH 5.0, 0.56 M Na+. | ||
Due to the hydrogen bonding interactions between hydrophobic PHEMA-g-(PCL-A) and hydrophilic PEG-U, the supramolecular brush copolymer micelles were unstable at acidic pH and high salt concentration. To evaluate their responsive ability, the micelles were treated with different pH buffer and salt concentration and the particle sizes were recorded by DLS measurements after half an hour. As shown in Fig. 6B, in a 0.12 M salt solution at pH 7.4, the diameter of micelles has a slight increase compared with that in deionized water, which indicates that a low salt concentration can hardly break the core–shell structures of the micelles. When the salt concentrations are increased to 0.56 M at pH 7.4, the size of the micelles increases from 108 nm to about 540 nm in half an hour. In the presence of high salt concentration, fast aggregation of the micelles is observed (Fig. 6C). On the other hand, when the pH is lowered from neutral to a value of 5.0, the particle size increases from 540 nm to roughly 700 nm (Fig. 6D). This can be explained by the increased protonation of the nitrogen atoms of the nucleobases and subsequent departure of hydrophilic PEG shells, leading to an increase in the particle size.
Drug loading and in vitro drug release
To better understand the potential of supramolecular brush copolymer micelles based on nucleobase recognition, DOX was selected as a model anticancer drug to determine their drug loading and release properties. The hydrophobic DOX was encapsulated into the hydrophobic inner cores of PHEMA-g-(PCL-A:U-PEG) micelles. When the feed ratio of copolymer to DOX was 10:1, the final DLC and DLE of supramolecular brush micelles were 7.0% and 70%, respectively. The supramolecular brush copolymer micelles exhibited high drug loading capacity and efficiency. In general, for linear polymers, the hydrophobic drug was surrounded in the core by individual polymer chains. However, both the inter- and intra-molecular hydrophobic interactions in brush polymers may contribute to the drug encapsulation, leading to improved drug loading capacity and efficiency. When the feed ratio of polymer to DOX was changed to 5:1, the DLC and DLE for the micelles of PHEMA-g-(PCL-A:U-PEG) were 4.3% and 86%. After drug loading, the size of micelles shows a slight decrease in average size, from 60 nm to 50 nm as measured by TEM (Fig. 5B). Correspondingly, the size and size distribution of DOX-loaded micelles by DLS measurement reveals that encapsulation of DOX into micelles decreases the diameters of micelles, and the Z-average diameters are 96 nm with a PDI of 0.11 (Fig. S3, ESI†).
The controlled drug releasing property of DOX-loaded PHEMA-g-(PCL-A:U-PEG) micelles was investigated under different pH and salt concentration using the dialysis method. 0.12 and 0.56 M sodium chloride concentrations were chosen to mimic the salt concentration in normal and tumor cells. As shown in Fig. 7, there is a slow release of about 4% of the incorporated DOX within 25 h in deionized water. In a 0.12 M salt solution at pH 7.4, about 20% DOX is released in 25 h. When the salt concentration reaches 0.56 M, a burst release of DOX from the micelles is observed and about 50% DOX is released in 25 h. High salt concentration leads to the dissociation of the hydrogen bonds, followed by a triggered fast release of the loaded DOX. When the drug-loaded micelles are dispersed in an acidic environment with a pH value of 5.0, however, the drug molecules entrapped in the supramolecular micelles will be quickly released with quick demicellization. In 0.12 M salt solution at pH 5.0, about 60% DOX is released in 25 h. When the salt concentration reaches 0.56 M at pH 5.0, over 70% DOX is burst-released in the first 6 h and up to 90% DOX is released in 25 h. From the above data, we can see that an acidic medium significantly accelerates DOX release from the micelles. The fast release of DOX from the DOX-loaded micelles in an acidic environment is likely due to the protonation of the amino group of the nucleobase and shedding of the micelle shell in acidic conditions. Another reason is the higher solubility of DOX in acidic media, so DOX could diffuse from micelles to media much faster in an acidic media than in neutral media. Therefore, a sufficiently high concentration of DOX can be achieved within a reasonably short period of time once the micelles are taken up by the tumor cells via endocytosis, thereby greatly enhancing the tumor-directed therapeutic efficacy.
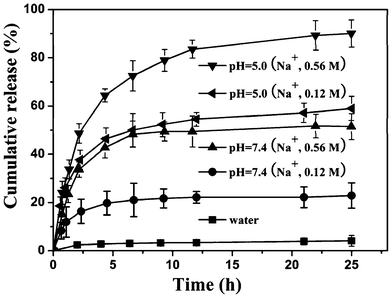 | ||
| Fig. 7 In vitro release profiles of DOX from PHEMA-g-(PCL-A:U-PEG) micelles at different pH values and Na+ concentrations at 37 °C. | ||
In vitro cytotoxicity of PHEMA-g-(PCL-A:U-PEG) micelles
The cytotoxicity of PHEMA-g-(PCL-A:U-PEG) micelles was evaluated by MTT assay using a mouse embryonic fibroblast NIH/3T3 cell line. The MTT assay is based on the ability of a mitochondrial dehydrogenation enzyme in viable cells to cleave the tetrazolium rings of the pale-yellow MTT and form formazan crystals with dark-blue color. Therefore, the number of surviving cells is directly proportional to the level of formed formazan.40Fig. 8 shows the cell viability after 24 h incubation with the micelles of PHEMA-g-(PCL-A:U-PEG) at different concentrations. The results demonstrate that no obvious cytotoxicity against NIH/3T3 cells is observed even when the concentration of copolymer micelles is as high as 500 μg mL−1. Apparently, these PHEMA-g-(PCL-A:U-PEG) micelles have low cytotoxicity to NIH/3T3 cells.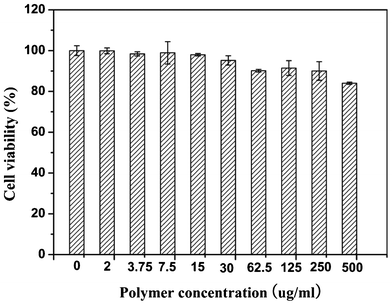 | ||
| Fig. 8 Cell viability of NIH/3T3 cells against PHEMA-g-(PCL-A:U-PEG) micelles after cultured for 24 h with different micelle concentrations. | ||
Cell internalization
The cellular uptake of DOX-loaded PHEMA-g-(PCL-A:U-PEG) micelles by Hela cells was analyzed by flow cytometry. DOX-loaded micelles with DOX concentration of 9 μg mL−1 were added to culture medium and the Hela cells were further cultured at 37 °C for the predetermined time intervals. The cellular uptake of PHEMA-g-(PCL-A:U-PEG) micelles is expressed as an increase in the fluorescent cell number. As shown in Fig. 9, the flow cytometric histograms of the pretreated cells incubated with DOX-loaded PHEMA-g-(PCL-A:U-PEG) micelles shift clearly in the direction of higher fluorescence intensity compared with that of the control cells. This result demonstrates effective internalization of DOX-loaded PHEMA-g-(PCL-A:U-PEG) micelles.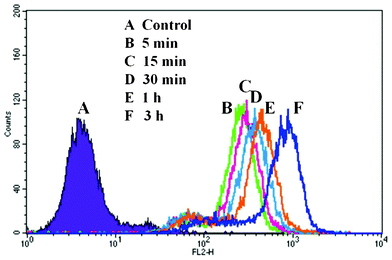 | ||
| Fig. 9 Flow cytometry histogram profiles of Hela cells that were incubated with self-assembled PHEMA-g-(PCL-A:U-PEG) micelles containing DOX for different time intervals. | ||
The intracellular drug release against Hela cells incubated with DOX-loaded PHEMA-g-(PCL-A:U-PEG) micelles was further studied by CLSM measurements. Hela cells were incubated with DOX-loaded PHEMA-g-(PCL-A:U-PEG) micelles (9 μg mL−1 of DOX) for 30 min. As a control, Hela cells were also incubated with the same dose of free DOX for 30 min. Then the nucleus was stained with Hoechst 33342. The intracellular fluorescence intensity and localization of DOX within the cells were studied by using the red autofluorescence from DOX and the blue fluorescence from Hoechst 33342. Fig. 10A and B show differential interference contrast (DIC), red DOX fluorescence, blue DAPI fluorescence, and merged images of the Hela cells pretreated by DOX-loaded micelles and free DOX, respectively. The DOX fluorescence in all cells is accumulated preferentially in the cytoplasm of the cells instead of the nucleus when the cells are cultured in the DOX-loaded micelles for 30 min. In contrast, the strong red fluorescence of DOX is mostly localized in the cell nucleus when cells are pretreated by free DOX for 30 min. Unlike the simple passive diffusion of small molecules through the membrane, the DOX-loaded PHEMA-g-(PCL-A:U-PEG) micelles may be taken up by a nonspecific endocytosis mechanism.
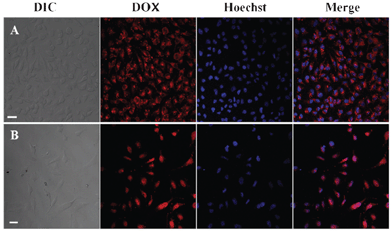 | ||
| Fig. 10 CLSM images of Hela cells (A) incubated with DOX-loaded PHEMA-g-(PCL-A:U-PEG) micelles for 30 min; (B) incubated with free DOX for 30 min. Cell nuclei were stained with Hoechst 33342; scale bars correspond to 30 μm in all images. | ||
In vitro cytotoxicity of drug-loaded micelles
The in vitro cytotoxicity of DOX-loaded PHEMA-g-(PCL-A:U-PEG) copolymer micelles compared with that of free DOX was evaluated by MTT assay against Hela cancer cells. The Hela cells were treated with the DOX-loaded PHEMA-g-(PCL-A:U-PEG) micelles and free DOX at different DOX doses from 0.05 to 2.5 μg mL−1. The Hela cell proliferation results are shown in Fig. 11. It was found that the loaded DOX dose required for 50% cellular growth inhibition (IC50) is 1.56 μg mL−1. This result demonstrates that DOX-loaded PHEMA-g-(PCL-A:U-PEG) micelles are able to enter the cells and produce the desired pharmacological action. On the other hand, the IC50 of free DOX is 0.97 μg mL−1, which exhibits higher inhibition compared with DOX-loaded micelles. It is well documented in previous literature that the free drug is more potent than the loaded drug.30,41 The lower cytotoxicity of DOX-loaded micelles may be attributed to the time consuming DOX release from micelles and delayed nuclear uptake in Hela cells.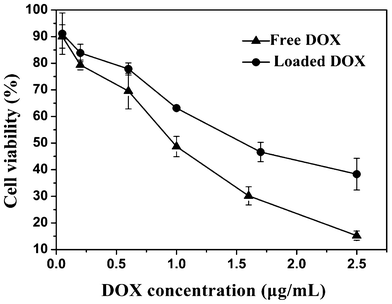 | ||
| Fig. 11 Cytotoxicity of free DOX and DOX-loaded PHEMA-g-(PCL-A:U-PEG) micelles to Hela cells after 48 h incubation (n = 3). | ||
Conclusions
In conclusion, we demonstrated that supramolecular micelles were easily self-assembled from supramolecular amphiphilic brush copolymer PHEMA-g-(PCL-A:U-PEG) acting as a dynamic carrier. A chemotherapeutic drug DOX could be efficiently encapsulated within the supramolecular micelles. The highest rate of DOX release from the copolymer micelles was observed in an environment at a low pH value and with a high level of salt concentration, which was analogous to pH and salt conditions in tumor cells. CLSM images indicated a fast internalization of the DOX-loaded micelles and efficient intracellular drug delivery. We envision that this supramolecular brush copolymer micelles system will lead to a new generation of stimuli-responsive drug delivery vehicles for intracellular delivery of a wide variety of biologically relevant molecules, and stimulate the development of unique and clinically applicable therapies.Acknowledgements
This work is sponsored by the National Basic Research Program (2009CB930400, 2012CB821500), National Natural Science Foundation of China (20974062), and China National Funds for Distinguished Young Scientists (21025417).References
- E. S. Lee, K. Na and Y. H. Bae, Nano Lett., 2005, 5, 325–329 CrossRef CAS.
- D. Schmaljohann, Adv. Drug Delivery Rev., 2006, 58, 1655–1670 CrossRef CAS.
- N. Rapoport, Prog. Polym. Sci., 2007, 32, 962–990 CrossRef CAS.
- S. Ganta, H. Devalapally, A. Shahiwala and M. Amiji, J. Controlled Release, 2008, 126, 187–204 CrossRef CAS.
- Y. F. Zhou, W. Huang, J. Y. Liu, X. Y. Zhu and D. Y. Yan, Adv. Mater., 2010, 22, 4567–4590 CrossRef CAS.
- L. J. Zhu, C. L. Tu, B. S. Zhu, Y. Su, Y. Pang, D. Y. Yan, J. L. Wu and X. Y. Zhu, Polym. Chem., 2011, 2, 1761–1768 RSC.
- C. Y. Zhang, Y. Q. Yang, T. X. Huang, B. Zhao, X. D. Guo, J. F. Wang and L. J. Zhang, Biomaterials, 2012, 33, 6273–6283 CrossRef CAS.
- J. Z. Du, X. J. Du, C. Q. Mao and J. Wang, J. Am. Chem. Soc., 2011, 133, 17560–17563 CrossRef CAS.
- S. Ganta, H. Devalapally, A. Shahiwala and M. Amiji, J. Controlled Release, 2008, 126, 187–204 CrossRef CAS.
- J. Jung, I. H. Lee, E. Lee, J. Park and S. Jon, Biomacromolecules, 2007, 8, 3401–3407 CrossRef CAS.
- E. R. Gillies and J. M. J. Fréchet, Chem. Commun., 2003, 1640–1641 RSC.
- E. R. Gillies and J. M. J. Fréchet, Bioconjugate Chem., 2005, 16, 361–368 CrossRef CAS.
- L. He, Y. Jiang, C. L. Tu, G. L. Li, B. S. Zhu, C. Y. Jin, Q. Zhu, D. Y. Yan and X. Y. Zhu, Chem. Commun., 2010, 46, 7569–7571 RSC.
- Y. Bae, S. Fukushima, A. Harada and K. Kataoka, Angew. Chem., Int. Ed., 2003, 42, 4640–4643 CrossRef CAS.
- C. C. Lee, E. R. Gillies, M. E. Fox, S. J. Guillaudeu, J. M. J. Frechet, E. E. Dy and F. C. Szoka, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16649–16654 CrossRef CAS.
- E. S. Lee, Z. Gao and Y. H. Bae, J. Controlled Release, 2008, 132, 164–170 CrossRef CAS.
- A. P. Griset, J. Walpole, R. Liu, A. Gaffey, Y. L. Colson and M. W. Grinstaff, J. Am. Chem. Soc., 2009, 131, 2469–2471 CrossRef CAS.
- I. Cameron, N. Smith, T. Pool and R. Sparks, Cancer Res., 1980, 40, 1493–1500 CAS.
- A. Luciani, Eur. J. Cell Biol., 2001, 80, 187–195 CrossRef CAS.
- L. Y. Qiu and Y. H. Bae, Pharm. Res., 2006, 23, 1–30 CrossRef CAS.
- Y. Geng, P. Dalhaimer, S. Cai, R. Tsai, M. Tewari, T. Minko and D. E. Discher, Nat. Nanotechnol., 2007, 2, 249–255 CrossRef CAS.
- L. Brunsveld, B. Folmer, E. Meijer and R. Sijbesma, Chem. Rev., 2001, 101, 4071–4097 CrossRef CAS.
- J. M. Lehn, Polym. Int., 2002, 51, 825–839 CrossRef CAS.
- W. Tao, Y. Liu, B. B. Jiang, S. R. Yu, W. Huang, Y. F. Zhou and D. Y. Yan, J. Am. Chem. Soc., 2012, 134, 762–764 CrossRef CAS.
- G. ten Brinke, J. Ruokolainen and O. Ikkala, Hydrogen-Bonded Polym., 2007, 207, 113–177 CrossRef CAS.
- M. Fathalla, C. M. Lawrence, N. Zhang, J. L. Sessler and J. Jayawickramarajah, Chem. Soc. Rev., 2009, 38, 1608–1620 RSC.
- P. Singh, J. Kumar, F. M. Toma, J. Raya, M. Prato, B. Fabre, S. Verma and A. Bianco, J. Am. Chem. Soc., 2009, 131, 13555–13562 CrossRef CAS.
- S. H. Kim, J. P. K. Tan, F. Nederberg, K. Fukushima, J. Colson, C. Yang, A. Nelson, Y.-Y. Yang and J. L. Hedrick, Biomaterials, 2010, 31, 8063–8071 CrossRef CAS.
- D. L. Wang, Y. Su, C. Y. Jin, B. S. Zhu, Y. Pang, L. J. Zhu, J. Y. Liu, C. L. Tu, D. Y. Yan and X. Y. Zhu, Biomacromolecules, 2011, 12, 1370–1379 CrossRef CAS.
- J. Z. Du, L. Y. Tang, W. J. Song, Y. Shi and J. Wang, Biomacromolecules, 2009, 10, 2169–2174 CrossRef CAS.
- W. Zhang, Y. Li, L. Liu, Q. Sun, X. Shuai, W. Zhu and Y. Chen, Biomacromolecules, 2010, 11, 1331–1338 CrossRef CAS.
- K. L. Beers, S. Boo, S. G. Gaynor and K. Matyjaszewski, Macromolecules, 1999, 32, 5772–5776 CrossRef CAS.
- M. Weisser, J. Käshammer, B. Menges, J. Matsumoto, F. Nakamura, K. Ijiro, M. Shimomura and S. Mittler, J. Am. Chem. Soc., 2000, 122, 87–95 CrossRef CAS.
- J. C. Kim, J. Jung, Y. Rho, M. Kim, W. Kwon, H. Kim, I. J. Kim, J. R. Kim and M. Ree, Biomacromolecules, 2011, 12, 2822–2833 CrossRef CAS.
- J. Weaver, I. Bannister, K. Robinson, X. Bories-Azeau, S. Armes, M. Smallridge and P. McKenna, Macromolecules, 2004, 37, 2395–2403 CrossRef CAS.
- J. K. Oh and K. Matyjaszewski, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 3787–3796 CrossRef CAS.
- A. S. Karikari, B. D. Mather and E. Timothy, Biomacromolecules, 2007, 8, 302–308 CrossRef CAS.
- A. Dong, T. Wan, S. Feng and D. Sun, J. Polym. Sci., Part B: Polym. Phys., 1999, 37, 2642–2650 CrossRef CAS.
- F. B. De Sousa, A. C. Lima, Â. M. L. Denadai, C. P. A. Anconi, W. B. De Almeida, W. T. G. Novato, H. F. Dos Santos, C. L. Drum, R. Langer and R. D. Sinisterra, Phys. Chem. Chem. Phys., 2012, 14, 1934–1944 RSC.
- J. Y. Liu, W. Huang, Y. Pang, X. Y. Zhu, Y. F. Zhou and D. Y. Yan, Langmuir, 2010, 26, 10585–10592 CrossRef CAS.
- X. T. Shuai, H. Ai, N. Nasongkla, S. Kim and J. Gao, J. Controlled Release, 2004, 98, 415–426 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra21923a |
| This journal is © The Royal Society of Chemistry 2012 |