Ultrafine nanoporous Cu–Pd alloys with superior catalytic activities towards electro-oxidation of methanol and ethanol in alkaline media
Zhonghua
Zhang
*,
Chi
Zhang
,
Junzhe
Sun
,
Tianyi
Kou
and
Changchun
Zhao
Key Laboratory for Liquid-Solid Structural Evolution and Processing of Materials (MOE), School of Materials Science and Engineering, Center for Advanced Energy Materials and Technology Research, Shandong University, Jingshi Road 17923, Jinan 250061, P.R. China. E-mail: zh_zhang@sdu.edu.cn; Fax: +86-531-88396978; Tel: +86-531-88396978
First published on 11th October 2012
Abstract
In this work, the dealloying of ternary Mg–Cu–Pd alloys and formation of nanoporous Cu–Pd alloys have been investigated using X-ray diffraction (XRD), scanning electron microscopy (SEM), transmission electron microscopy (TEM), high resolution TEM (HRTEM), energy dispersive X-ray (EDX) analysis and electrochemical measurements. The results show that the Pd addition has a significant influence on the phase constitution and dealloying process of the rapidly solidified Mg–Cu–Pd alloys. Ultrafine nanoporous Cu–Pd alloy nanostructures with ligaments/channels of less than 10 nm can be obtained in the as-dealloyed samples. The dealloying mechanism and formation of ultrafine nanoporous structures have been rationalized by electrochemical activity measurements and surface diffusion of Cu/Pd adatoms. These ultrafine nanoporous Cu–Pd alloys exhibit high specific surface areas and superior electrocatalytic performance towards electro-oxidation of methanol and ethanol in alkaline media. The present findings provide a facile dealloying route to fabricate nanoporous Cu–Pd alloy electrocatalysts for applications in direct alcohol fuel cells.
Introduction
Nanoporous metals with high specific surface areas and unique bicontinuous ligament-channel structures show great potential in many fields such as catalysis, sensors, actuators, fuel cells, and so on.1–4 Dealloying, which has historically been considered as a concept of corrosion, has been developed to be an important method for fabricating nanoporous metals with a three-dimensional bicontinuous interpenetrating ligament-channel structure.5,6 Alloy systems including Au–Ag,1,2,4–6 Al–Cu,7 Cu–Zr,8 Cu–Au,9 Pt–Si,10etc, have been explored to produce the corresponding nanoporous metals through the dealloying process. Except for solid solution alloys like Ag–Au and Cu–Mn,1,2,4–6,11 other alloy systems (intermetallic compounds,8,9,12 quasicrystalline13 and even amorphous alloys10,14) have also been reported to form nanoporous structures by dealloying.For applications in fuel cells, great efforts have been dedicated towards synthesis of nanoporous metals or alloys with designed compositions and nanostructures in order to achieve better precious-metal utilization and performance. Through the combination of dealloying and the Cu-under-potential-deposition (UPD)-mediated process, Wang et al.4 successfully fabricated a new type of nanostructured electro-catalyst that simultaneously fulfils three key requirements for a good practical formic acid electro-oxidation catalyst: ultralow Pt loading, great tolerance to poisoning, and high stability. Using porous anodic aluminium oxide (AAO) template-assisted electrodeposition and mild dealloying techniques, Liu et al.15–17 synthesized nanoporous Pt-based alloy (PtCo,15 PtNi,16 PtCuCoNi17) nanowires with superior electro-catalytic performance. Shui et al.18 used electrospinning and chemical dealloying techniques to make long, thin and nanoporous Pt–Fe alloy nanowires which show high catalytic property for oxygen reduction reaction (ORR). Based on a combination of room-temperature dealloying and galvanic-replacement reaction, Xu et al.19 produced nanotubular mesoporous bimetallic nanocatalysts (Pt/Cu and Pd/Cu) with enhanced electrocatalytic properties. Recently, it has been found that the addition of element(s) with lower diffusivity (such as Pt and Pd) into precursor alloys can effectively inhibit surface diffusion of more noble adatoms during dealloying and contribute to the formation of ultrafine nanoporous alloys with excellent electrocatalytic activities.20–22
Compared to Pt-based catalysts, Pd is much cheaper and possesses potential advantages in applications to direct alcohol fuel cells (DAFCs) and direct formic acid fuel cells (DFAFCs).23,24 Therefore, lots of Pd catalysts with various morphologies and structures have been synthesized, such as nanoparticles,25 nanoplate arrays,26 nanotrees,27 nanowires,28 nanoflowers,29etc. In our former work, nanoporous palladium (NPPd) with high electrocatalytic activity was fabricated by dealloying Al–Pd alloys.30,31 Most recently, nanoporous Pt-based (PtCo, PtFe and PtNi) alloy catalysts with enhanced electrocatalytic performance were produced by dealloying Al-based ternary precursors in alkaline media.32–35 In contrast, however, less information is available on nanoporous Pd-based alloy catalysts fabricated by dealloying.36 Here, we aim at investigating the dealloying process of rapidly solidified Mg–Cu–Pd alloys and formation of ultrafine nanoporous Cu–Pd alloys. The electrocatalytic activities of these nanoporous alloys have also been evaluated.
Experimental section
Mg–Cu–Pd alloys with nominal compositions of Mg50Cu45Pd5 and Mg50Cu40Pd10 (at.%) were prepared from pure elements (purity: 99.9 wt. %) in a quartz crucible using a high frequency induction furnace in an argon atmosphere. Using a single roller melt spinning apparatus, the prealloyed ingots were rapidly solidified into ribbons with 20–50 μm in thickness. The precursor alloy ribbons were dealloyed in a 5 wt. % HCl aqueous solution firstly at room temperature and then at 90 ± 5 °C until no visible bubbles emerged. The as-dealloyed samples were then rinsed using distilled water and dehydrated alcohol.The phases present in the precursor alloys and as-dealloyed samples were identified using an X-ray diffractometer (XRD, Hitachi Rigaku D/max-RB) with Cu-Kα radiation. The microstructure of the as-dealloyed samples was characterized using SEM (LEO 1530VP in an Inlens mode), transmission electron microscopy (TEM, Philips CM 20), and high-resolution TEM (HRTEM, FEI Tecnai G2). The chemical compositions of the as-dealloyed samples were determined by an energy-dispersive X-ray (EDX) analyzer which was attached to SEM. In addition, scanning transmission electron microscopy (STEM) images and nanobeam-EDX (NB-EDX) spectra were also obtained by the FEI Tecnai G2 microscope under high-angle annular dark filed (HAADF) mode. For one composition, at least 3–5 EDX experiments were carried out and an average value is given in the results.
All electrochemical measurements were performed in a standard three-electrode cell using a LK 2005A potentiostat. Open-circuit measurements and potentiodynamic anodic polarization were used to evaluate the electrochemical activities of the rapidly solidified Mg–Cu–Pd precursor alloys. The Mg–Cu–Pd alloy ribbons were directly used as the work electrode. The Ag/AgCl electrode (in a saturated KCl solution) was used as the reference electrode, along with a platinum plate as the counter electrode. A 0.2 M NaCl aqueous solution was used as the electrolyte. In order to evaluate the electrocatalytic activities of the as-dealloyed Mg–Cu–Pd alloys, cyclic voltammetry (CV) measurements were performed. The as-dealloyed Mg–Cu–Pd electrodes were prepared as follow: 4 mg as-dealloyed samples, 4 mg carbon powder, 300 μL isopropanol, and 100 μL Nafion solution (0.5 wt. %) were ultrasonically mixed. 5 μL catalyst ink was placed on a polished glassy carbon (GC) electrode with a diameter of 4 mm being used as the work electrode. The counter electrode was a platinum plate, and a saturated calomel electrode (SCE) or a Hg/HgO (MMO) was used as the reference electrode. The voltammetric behavior of the as-dealloyed samples was characterized in a 1.0 M H2SO4 solution. Electrocatalytic activities were measured in solutions of 1.0 M KOH + 0.5 M methanol and 1.0 M KOH + 1.0 M ethanol. Prior to electrochemical measurements, the electrolytes were deoxygenated by bubbling with N2 for 30 min. All electrochemical experiments were carried out at room temperature. The current densities were normalized by the geometric area of each electrode (jgeom.) as well as by the mass of Pd (jmass).
Results and discussion
Dealloying of Mg–Cu–Pd alloys and formation of nanoporous structures
Fig. 1 shows the XRD patterns of the rapidly solidified Mg–Cu–Pd alloys and as-dealloyed samples. For comparison, the XRD patterns of the rapidly solidified and as-dealloyed Mg50Cu50 alloys are also presented in Fig. 1. The rapidly solidified Mg50Cu50 alloy is composed of two phases: Mg2Cu (PDF No. 13-0504) and MgCu2 (PDF No. 65-3033) intermetallic compounds.37 The rapidly solidified Mg50Cu45Pd5 alloy mainly consists of Mg2Cu and MgCu2 (Fig. 1a). Thus, it is reasonable to assume that Pd exists in these two intermetallic phases in the form of a solid solution. The addition of 10 at.% Pd markedly alters the phase constitution of the rapidly solidified Mg50Cu40Pd10 alloy, which is comprised of MgPd (PDF No. 65-6763) and MgCu2 (Fig. 1a).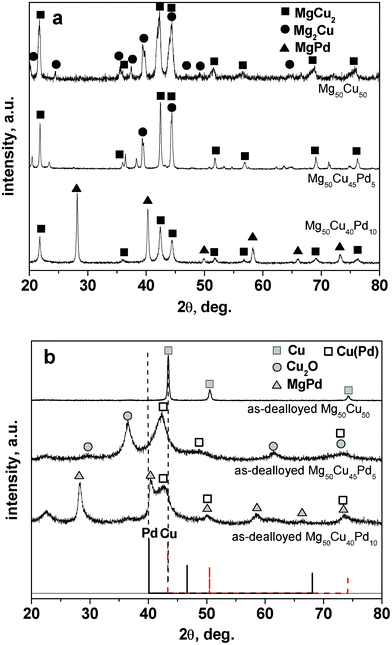 | ||
| Fig. 1 XRD patterns of (a) the rapidly solidified Mg50Cu50-xPdx (x = 0, 5, 10) precursor alloys and (b) the as-dealloyed samples in the 5% HCl solution. | ||
It is clear that only a face centered cubic (f.c.c.) Cu phase can be identified in the as-dealloyed Mg50Cu50 alloy (Fig. 1b). On the XRD pattern of the as-dealloyed Mg50Cu45Pd5 alloy, there exist broad diffraction peaks (at 42.25°, 48.74° and 72.98°) which are located between the standard lines of f.c.c. Cu and Pd, indicating the formation of Cu(Pd) solid solution alloy (Fig. 1b). And, these peaks correspond to the (111), (200) and (220) reflections of the f.c.c. phase. In addition, Cu oxide (Cu2O) can also be identified in the as-dealloyed Mg50Cu45Pd5 sample. In fact, the oxidation of Cu is inevitable in the formation of RANEY® copper38 and in the dealloying of Al–Cu–Fe quasicrystalline alloys,13 especially for fine Cu nanostructures. Similarly, the f.c.c. Cu(Pd) solid solution phase also appears in the as-dealloyed Mg50Cu40Pd10 alloy (Fig. 1b). However, the MgPd phase cannot be dealloyed in the 5 wt. % HCl solution and remains in the as-dealloyed sample. The present XRD results demonstrate that the rapidly solidified Mg50Cu45Pd5 alloy can be fully dealloyed in the HCl solution but the Mg50Cu40Pd10 alloy cannot be completely dealloyed.
Fig. 2 shows the section-view SEM images of the as-dealloyed Mg–Cu–Pd samples. A typical nanoporous structure can be observed in the as-dealloyed Mg50Cu45Pd5 alloy (Fig. 2a). At a higher magnification, the ligaments of less than 10 nm can be seen in the nanoporous structure (Fig. 2b). The EDX results show that the as-dealloyed sample is mainly composed of Cu and Pd, and the composition was determined as 81.8 at.% Cu, 12.3 at.% Pd and 5.9 at.% Mg. A typical EDX spectrum is shown in Fig. 2a as an inset. As shown in Fig. 2c, the microstructure of the as-dealloyed Mg50Cu40Pd10 sample is quite different from that of the as-dealloyed Mg50Cu45Pd5 alloy, and the nanoporous structure cannot be clearly observed. Some ligaments of less than 10 nm are present in some zones of the higher-magnification image (Fig. 2d). The EDX analysis demonstrates that an appreciable amount of Mg can be detected in the as-dealloyed sample and its composition was determined to be 50.8 at.% Cu, 18.3 at.% Pd and 30.9 at.% Mg. A typical EDX spectrum is shown in Fig. 2c as an inset. The present EDX analysis of the as-dealloyed Mg–Cu–Pd samples is in good agreement with the XRD results shown in Fig. 1b.
 | ||
| Fig. 2 SEM images showing the microstructure of the as-dealloyed Mg–Cu–Pd alloys ((a,b) Mg50Cu45Pd5, (c,d) Mg50Cu40Pd10) in the 5 wt.% HCl solution. Insets in (a) and (c) show the corresponding EDX spectra. | ||
In order to clarify the nanostructure, TEM and selected-area electron diffraction (SAED) were used to document the microstructure of the as-dealloyed Mg50Cu40Pd10 alloy (Fig. 3). As shown in Fig. 3a, it is interesting to note that two different zones can be clearly observed in the as-dealloyed Mg50Cu40Pd10 alloy: solid zones (zone 1) and nanoporous zones (zone 2). Moreover, the nanoporous zones exhibit a bicontinuous interpenetrating ligament-channel structure, and the length scale of ligaments/channels is ∼7 nm (Fig. 3b). The SAED analysis confirms that the solid zones correspond to the MgPd phase, and a pattern of MgPd [111] zone axis is presented in Fig. 3c. In contrast, the nanoporous zones are nanocrystalline and a set of diffraction rings can be clearly seen in the corresponding SAED pattern (Fig. 3d). In combination with the XRD results, these rings can be indexed as (111), (200), (220) and (311) reflections of the f.c.c. Cu(Pd) phase. In addition, the HRTEM observation further verifies the ultrafine ligament-channel structure of the nanoporous zones (Fig. 3e). Some nanocrystals with sizes of 5–8 nm can be clearly seen.
 | ||
| Fig. 3 (a,b) TEM and (e) HRTEM images showing the microstructure of the as-dealloyed Mg50Cu40Pd10 alloy. (c,d) SAED patterns corresponding to the different zones in (a). | ||
To accurately determine the chemical compositions of the different zones in the as-dealloyed Mg50Cu40Pd10 alloy, STEM observation was performed under the HAADF mode and one typical image is shown in Fig. 4a. Both the solid and nanoporous zones can be clearly observed in the z-contrast STEM image. Typical NB-EDX spectra are shown in Fig. 4b and c for the solid (zone 1) and nanoporous (zone 2) zones, respectively. It is obvious that the composition of the solid zones is quite different from that of the nanoporous zones. The composition of the solid zones was determined to be 41.9 at.% Mg, 26.6 at.% Cu and 31.5 at.% Pd, while that of the nanoporous zones is 64.2 at.% Cu, 31.3 at.% Pd and 4.5 at.% Mg. In combination with the XRD and SAED results, the solid zones correspond to the MgPd phase and Cu atoms occupy some lattice sites substituting for Pd. Therefore, the phase in the solid zones can be designated as Mg(Pd,Cu). The nanoporous zones are the Cu(Pd) solid solution phase with minor residual Mg.
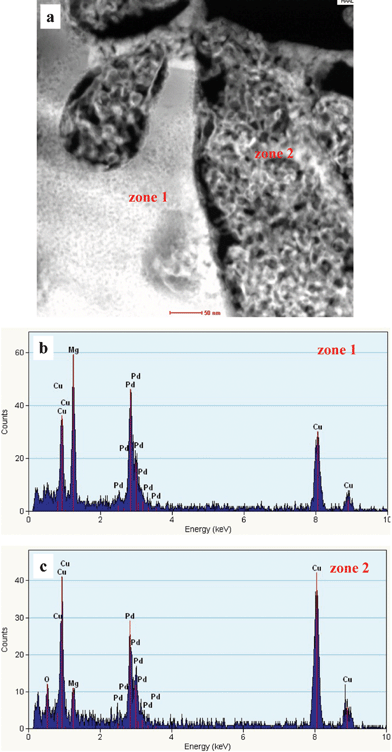 | ||
| Fig. 4 (a) HAADF-STEM image showing the microstructure of the as-dealloyed Mg50Cu40Pd10 alloy, and (b,c) corresponding NB-EDX spectra. | ||
As mentioned above, the Pd addition has an important influence on the dealloying process of the rapidly solidified Mg–Cu–Pd alloys. In the former work, we have found that both Mg2Cu and MgCu2 can be dealloyed in the HCl solution.37 Similarly to the Mg50Cu50 alloy composed of Mg2Cu and MgCu2, the rapidly solidified Mg50Cu45Pd5 alloy can be fully dealloyed in the HCl solution due to the selective dissolution of Mg. The phase constitution of the rapidly solidified Mg50Cu40Pd10 alloy is different from that of the Mg50Cu50 and Mg50Cu45Pd5 alloys (Fig. 1a). The Mg(Pd,Cu) phase in the Mg50Cu40Pd10 alloy is inert in the HCl solution and is retained in the as-dealloyed sample (solid zones, Fig. 3a). Meanwhile, the dealloying of MgCu2 (containing Pd) contributes to the formation of the Cu(Pd) alloy (nanoporous zones, Fig. 3a). Therefore, the dealloying of the Mg–Cu–Pd precursor alloys becomes more difficult with increasing Pd content. Eventually, this dealloying tendency can be reflected by the electrochemical activity of the precursor alloys. In order to avoid the interference by chemical corrosion, the electrochemical activity measurements were performed in a 0.2 M NaCl solution. Fig. 5 shows the open-circuit potential vs. time and potentiodynamic polarization curves of the rapidly solidified Mg50Cu50-xPdx (x = 0, 5, 10) precursor alloys. The average open-circuit potentials were determined to be −0.94, −0.84 and −0.76 V vs. Ag/AgCl for the Mg50Cu50, Mg50Cu45Pd5 and Mg50Cu40Pd10 alloys, respectively. So far, different methods have been proposed to define the critical potential (Ecrit) for the onset of bulk dealloying of an alloy.39–41 Here, the potential corresponding to the current density of 1 mA cm−2 was taken as Ecrit. From the potentiodynamic polarization curves, the Ecrit values were determined to be −0.87, −0.71 and −0.49 V vs. Ag/AgCl for the Mg50Cu50, Mg50Cu45Pd5 and Mg50Cu40Pd10 alloys, respectively. It is obvious that both the open-circuit and critical potentials increase with increasing Pd content in the Mg–Cu–Pd precursor alloys, which is consistent with their dealloying behaviors in the HCl solution under free corrosion conditions.
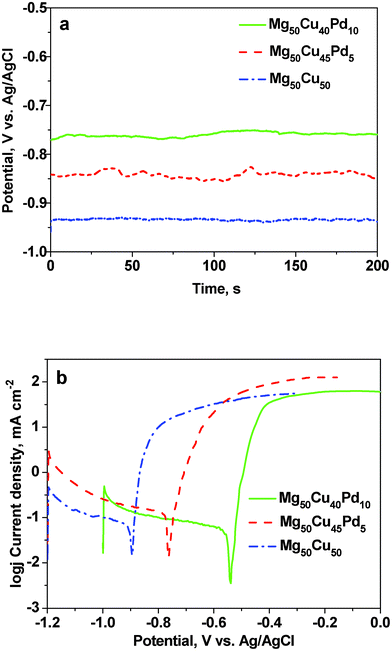 | ||
| Fig. 5 (a) Open-circuit potential vs. time and (b) potentiodynamic polarization curves of the rapidly solidified Mg50Cu50-xPdx (x = 0, 5, 10) precursor alloys. | ||
Besides, the Pd addition significantly refines the size of ligaments/channels in the as-dealloyed Mg50Cu45Pd5 and Mg50Cu40Pd10 samples (Fig. 2 and 3). It has been shown experimentally that surface diffusion of more noble elements along alloy/solution interfaces during dealloying has a significant influence on the length scale of ligaments/channels.5,6 Snyder et al.42 have argued that the additives possessing much slower surface diffusion rates will pin the mobile Au step edges and ultimately stabilize them, reducing the scale of porosity. During the dealloying of Mg60Ag32Pd8, the diffusion of Pd has a serious pinning effect on that of Ag, resulting in the formation of ultrafine ligaments/channels of as small as ∼5 nm in the nanoporous Ag–Pd alloy.43 In electrolytes, the surface mass transfer diffusion coefficients of Pd and Cu are of the order of magnitude of 10−16 and 10−10 cm2 s−1 respectively.44,45 Therefore, the diffusion rate of Pd is much slower than that of Cu. Kaiser et al.46 reported that alloy diffusivity would greatly influence the dealloying kinetics and corrosion morphology for the Cu–Pd alloy. In the present work, the active Mg atoms continuously dissolve into the HCl solution during dealloying, and then the sites that were once occupied by Mg atoms turn to vacancies. The released Cu and Pd atoms diffuse and re-organize into the nanoporous structure. Furthermore, Pd with lower diffusivity has a significant prohibition effect on the surface diffusion of Cu adatoms. Thus, ultrafine nanoporous structures with ligaments/channels of less than 10 nm can be obtained in the as-dealloyed Mg–Cu–Pd samples.
Electrocatalytic activities of as-dealloyed Mg–Cu–Pd alloys
The surface state of the as-dealloyed Mg–Cu–Pd samples was first detected in a 1.0 M H2SO4 solution by CV measurements (Fig. 6). As shown in Fig. 6a, during the first potential cycling, two broad oxidation peaks can be observed in the forward scan. And the associated charge is markedly larger than that in the back scan, indicating the occurrence of irreversible dissolution reaction in the forward scan. It is reasonable to assume that these broad oxidation peaks are mainly caused by dissolution of Cu and residual Mg in the as-dealloyed Mg50Cu45Pd5 electrode. In the subsequent cycles, the CV profiles gradually stabilize and behave similarly to the Pd-based electrode. For the 10th cycle, the CV curve exhibits a typical profile for Pd nanostructures.25–31 Although the 1st CV profile is different from that of the as-dealloyed Mg50Cu45Pd5 electrode, a similar CV evolution process can also be observed for the as-dealloyed Mg50Cu40Pd10 electrode (Fig. 6b). The dissolution of Mg in the Mg(Pd,Cu) phase may be involved in the initial cycles. Moreover, the final CV profile (10th cycle) of the as-dealloyed Mg50Cu40Pd10 electrode is similar to that of the as-dealloyed Mg50Cu45Pd5 electrode.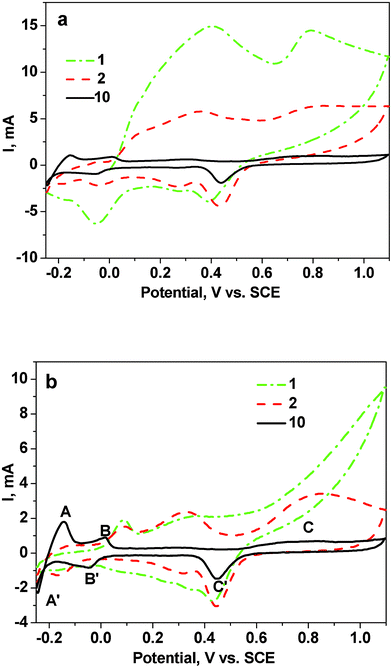 | ||
| Fig. 6 CV curves for the as-dealloyed Mg–Cu–Pd electrodes ((a) Mg50Cu45Pd5 and (b) Mg50Cu40Pd10) in a 1.0 M H2SO4 aqueous solution. The scanning rate was 50 mV s−1. | ||
Here, we take the final CV curve of the as-dealloyed Mg50Cu40Pd10 electrode as an example to analyze the electrochemical process during potential sweep. The CV curve shows two pairs of well-separated peaks between −0.25 and 0.05 V vs. SCE (A, A′, B and B′, Fig. 6b) related to the hydrogen adsorption–desorption process.47 In addition, the broad oxidation current peak (C) is ascribed to the formation of Pd surface oxide (PdO) and the corresponding peak (C′) in the back scan is caused by subsequent reduction of the oxide.48,49 The reduction peak (C′) occurs at 0.44 and 0.45 V vs. SCE for the as-dealloyed Mg50Cu45Pd5 and Mg50Cu40Pd10 electrodes respectively, slightly lower than that (0.46 V vs. SCE30) of the NPPd electrode. In some cases, the oxygen adsorption method is applicable to evaluating electrochemically active surface areas (EASAs) of metals (e.g. Au and Pd) with good affinity for oxygen.50 By integration of the cathodic peaks corresponding to reduction of the Pd oxide monolayer that formed in the previous anodic scan, the mass-normalized EASAs were calculated to be 71.9 and 39.5 m2 g−1 (Pd) for the as-dealloyed Mg50Cu45Pd5 and Mg50Cu40Pd10 electrodes respectively (Table 1). These values are much higher than that of NPPd obtained by dealloying Al–Pd alloys (20.5 m2 g−1)31 or Pd30Ni50P20 metallic glass (∼13 m2 g−1).14
| Sample | EASA (m2 g−1 Pd) | methanol | ethanol | ||||
|---|---|---|---|---|---|---|---|
| E p (V vs. MMO) | j p geom. (mA cm−2) | j p mass (mA mg−1 Pd) | E p (V vs. MMO) | j p geom. (mA cm−2) | j p mass (mA mg−1 Pd) | ||
| As-dealloyed Mg50Cu45Pd5 | 71.9 | −0.04 | 64.4 | 808.9 | −0.04 | 119.8 | 1504.7 |
| As-dealloyed Mg50Cu40Pd10 | 39.5 | −0.04 | 86.7 | 660.0 | −0.02 | 162.1 | 1233.9 |
| NPPd | 20.5 | −0.04 | 88.9 | 223.5 | −0.05 | 131.0 | 329.1 |
Fig. 7 shows the CV curves for the as-dealloyed Mg–Cu–Pd electrodes in a solution of 1.0 M KOH + 0.5 M methanol. Both electrodes show significantly high catalytic activity towards the electro-oxidation of methanol. In the forward scan, the peak potentials of both electrodes are comparable to that of the NPPd electrode (Table 1). The oxidation peak located in the forward scan is correlated with the oxidation of freshly chemisorbed methanol molecules, and is normally used to evaluate the catalytic activity of electrocatalysts.51 The anodic peak current in the reverse scan is connected with the oxidation of intermediate species adsorbed on Pd. The anodic peak current density (normalized by the geometric area of each electrode, jpgeom.) is 64.4 and 86.7 mA cm−2 for the as-dealloyed Mg50Cu45Pd5 and Mg50Cu40Pd10 electrodes respectively, lower than that (88.9 mA cm−231) of the NPPd electrode (Fig. 7 and Table 1). However, the mass activity (normalized by the mass of Pd on the GC electrode, jpmass) is 808.9 and 660.0 mA mg−1 Pd for the as-dealloyed Mg50Cu45Pd5 and Mg50Cu40Pd10 electrodes respectively, which is 3.6 and 3.0 times that (223.5 mA mg−131) of the NPPd electrode. Yin et al.29 have synthesized porous Pd nanoflowers with an average size of 50 nm, which exhibit a mass activity of ∼43 mA mg−1 for methanol electrooxidation in the KOH solution. It is obvious that the electrocatalytic performance of the present nanoporous Cu–Pd alloys is much better than that of the Pd nanoflowers. Jia et al.26 have reported an anodic peak current density of 137 mA cm−2 for Pd nanoplate array in the KOH + methanol solution. Considering the lower content of Pd in the as-dealloyed samples (20 and 33 wt.% Pd in the as-dealloyed Mg50Cu45Pd5 and Mg50Cu40Pd10 samples respectively), the electrocatalytic activities of the present electrocatalysts are still superior to those of the Pd nanoplate array.
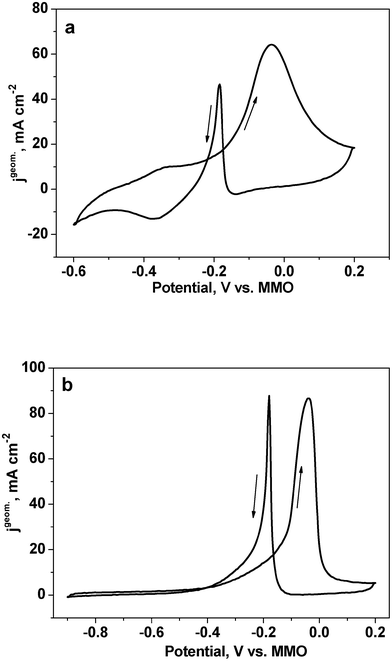 | ||
| Fig. 7 CV curves for the as-dealloyed Mg–Cu–Pd ((a) Mg50Cu45Pd5 and (b) Mg50Cu40Pd10) electrodes in the electrolyte of 1.0 M KOH + 0.5 M methanol. The scan rate of potential was 10 mV s−1. | ||
The electrocatalytic activities of the as-dealloyed Mg–Cu–Pd electrodes for the oxidation of ethanol were also characterized by CV measurements (Fig. 8). It is not unexpected that the as-dealloyed Mg–Cu–Pd electrodes show superior electrocatalytic activities towards the ethanol oxidation process. In the forward scan, the anodic peak potential for the as-dealloyed Mg–Cu–Pd electrodes is comparable to that of the NPPd electrode (Table 1). And the anodic peak current density (jpgeom.) of the as-dealloyed Mg–Cu–Pd electrodes attains 119.8 mA cm−2 (Mg50Cu45Pd5) and 162.1 mA cm−2 (Mg50Cu40Pd10), also comparable to that (131.0 mA cm−230) of the NPPd electrode. However, the mass activity (jpmass) reaches 1504.7 and 1233.9 mA mg−1 Pd for the as-dealloyed Mg50Cu45Pd5 and Mg50Cu40Pd10 electrodes respectively, which is 4.6 and 3.7 times that (329.1 mA mg−130) of the NPPd electrode (Table 1). Xu et al.28 have fabricated highly ordered Pd nanowire arrays which show a good electrocatalytic activity (peak current density of 74 mA cm−2) for ethanol oxidation in the KOH solution. Obviously, the present nanoporous Cu–Pd alloys show better electrocatalytic activities than the Pd nanowire arrays.
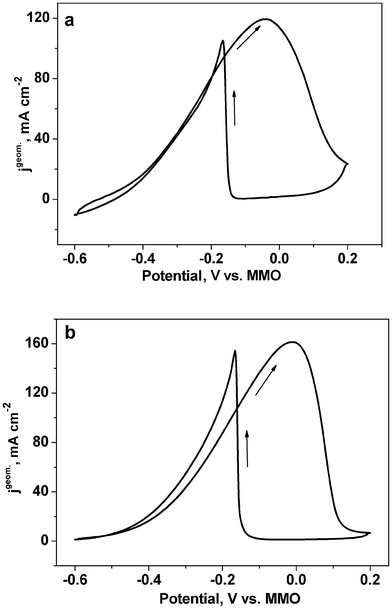 | ||
| Fig. 8 CV curves for the as-dealloyed Mg–Cu–Pd ((a) Mg50Cu45Pd5 and (b) Mg50Cu40Pd10) electrodes in the electrolyte of 1.0 M KOH + 1.0 M ethanol. The scan rate of potential was 10 mV s−1. | ||
The above results demonstrate that the as-dealloyed Mg–Cu–Pd alloys exhibit superior electrocatalytic activities towards the oxidation of methanol and ethanol in the alkaline solutions, even much higher than those of the NPPd sample (taking mass activity into consideration). The reasons for their enhanced catalytic activities can be suggested as follows. First of all, the Pd doping in the precursor alloys plays a dominant role in the formation of the ultrafine nanoporous structure (Fig. 1–4) and the excellent electrocatalytic activities of the as-dealloyed samples are due to the intrinsic catalytic nature of metallic Pd. Moreover, during potential cycling (Fig. 6), the dissolution of Cu and Mg may induce surface rearrangements, generating Pd-rich surfaces with a nanosized Pd–Cu alloy core.52 The sublayer Cu atoms may provide an electronic modification for the topmost Pd layer by a strain effect due to lattice mismatch and alloying effect from the formation of hetero-atom bond.53,54 Density function theory (DFT) calculations have shown that the d-band center of Pd shifts from −2.638 to −1.843 eV by the incorporation of Cu (from 0 to 50 at.%),55 which will weaken the adsorption of CO on Pd surface.56 The unique surface structure and the variation of the d-band center should be responsible for the greatly enhanced electrocatalytic activities of the as-dealloyed Mg–Cu–Pd alloys. In addition, the ultrafine nanoporous structure can provide much higher EASAs and the interconnected interstices and channels extending in all three dimensions can allow unblocked transport of medium molecules and electrons, which also contributes to the superior electrocatalytic activity of as-dealloyed Mg–Cu–Pd alloys. It should be noted that the as-dealloyed Mg50Cu45Pd5 alloy shows better catalytic activity (jpmass) than that of Mg50Cu40Pd10. The un-dealloyed Mg(Pd,Cu) phase in the latter should be the main reason for this.
Summary and conclusions
In summary, the Pd addition has a significant influence on the phase constitution and dealloying process of the rapidly solidified Mg–Cu–Pd alloys. The rapidly solidified Mg50Cu45Pd5 alloy can be fully dealloyed in a HCl solution, resulting in the formation of ultrafine nanoporous Cu–Pd alloy with ligaments/channels of less than 10 nm. The MgCu2 phase containing Pd in the Mg50Cu40Pd10 alloy can be dealloyed to form ultrafine nanoporous Cu–Pd zones with a ligament/channel size of ∼7 nm, while the Mg(Pd,Cu) phase cannot be dealloyed and is retained in the resultant sample. With increasing Pd content, the dealloying of the Mg–Cu–Pd alloys becomes more difficult, which is associated with their electrochemical activity (open-circuit potentials and critical potentials). The addition of Pd with lower surface diffusivity has a significant prohibition effect on surface diffusion of Cu adatoms during dealloying, leading to the formation of ultrafine nanoporous Cu–Pd alloy nanostructures. The nanoporous Cu–Pd alloys possess much higher specific surface areas and the mass-normalized EASAs reach 71.9 and 39.5 m2 g−1 (Pd) for the as-dealloyed Mg50Cu45Pd5 and Mg50Cu40Pd10 electrodes respectively. Moreover, these nanoporous Cu–Pd alloys exhibit superior electrocatalytic activities towards oxidation of methanol and ethanol in alkaline media, which are even much better than those of pure nanoporous Pd (NPPd). Based upon alloy design of ternary Mg-based precursors, the dealloying is a facile and effective routine to fabricate ultrafine nanoporous Cu–Pd alloys for anode catalysts of DAFCs.Acknowledgements
The authors gratefully acknowledge financial support by the National Basic Research Program of China (973, 2012CB932800), Program for New Century Excellent Talents in University (MOE), Independent Innovation Foundation of Shandong University (2010JQ015) and National Natural Science Foundation of China (50971079). The experimental assistance from Ruhr University at Bochum (Germany) is acknowledged. Z. H. Zhang acknowledges the support from the Alexander von Humboldt Foundation (Germany).References
- C. X. Xu, J. X. Su, X. H. Xu, P. P. Liu, H. J. Zhao, F. Tian and Y. Ding, J. Am. Chem. Soc., 2007, 129, 42 CrossRef CAS.
- J. Biener, A. Wittstock, L. A. Zepeda-Ruiz, M. M. Biener, V. Zielasek, D. Kramer, R. N. Viswanath, J. Weissmüller, M. Bäumer and A. V. Hamza, Nat. Mater., 2009, 8, 47 CrossRef CAS.
- H. J. Jin, X. L. Wang, S. Parida, K. Wang, M. Seo and J. Weissmüller, Nano Lett., 2010, 10, 187 CrossRef CAS.
- R. Wang, C. Wang, W. Cai and Y. Ding, Adv. Mater., 2010, 22, 1845 CrossRef CAS.
- A. J. Forty, Nature, 1979, 282, 597 CrossRef CAS.
- J. Erlebacher, M. J. Aziz, A. Karma, N. Dimitrov and K. Sieradzki, Nature, 2001, 410, 450 CrossRef CAS.
- Z. Qi, C. C. Zhao, X. G. Wang, J. K. Lin, W. Shao, Z. H. Zhang and X. F. Bian, J. Phys. Chem. C, 2009, 113, 6694 CAS.
- H. B. Lu, Y. Li and F. H. Wang, Scr. Mater., 2007, 56, 165 CrossRef CAS.
- F. U. Renner, A. Stierle, H. Dosch, D. M. Kolb, T. L. Lee and J. Zegenhagen, Nature, 2006, 439, 707 CrossRef CAS.
- J. C. Thorp, K. Sieradzki, L. Tang, P. A. Crozier, A. Misra, M. Nastasi, D. Mitlin and S. T. Picraux, Appl. Phys. Lett., 2006, 88, 033110 CrossRef.
- J. R. Hayes, A. M. Hodge, J. Biener, A. V. Hamza and K. J. Sieradzki, J. Mater. Res., 2006, 21, 2611 CrossRef CAS.
- Z. H. Zhang, Y. Wang, Z. Qi, W. H. Zhang, J. Y. Qin and J. Frenzel, J. Phys. Chem. C, 2009, 113, 12629 CAS.
- Z. Qi, Y. Z. Gong, C. Zhang, J. L. Xu, X. G. Wang, C. C. Zhao, H. Ji and Z. H. Zhang, J. Mater. Chem., 2011, 21, 9716 RSC.
- J. S. Yu, Y. Ding, C. X. Xu, A. Inoue, T. Sakurai and M. W. Chen, Chem. Mater., 2008, 20, 4548 CrossRef CAS.
- L. F. Liu, E. Pippel, R. Scholz and U. Gösele, Nano Lett., 2009, 9, 4352 CrossRef CAS.
- L. F. Liu, R. Scholz, E. Pippel and U. Gösele, J. Mater. Chem., 2010, 20, 5621 RSC.
- L. F. Liu and E. Pippel, Angew. Chem., Int. Ed., 2011, 50, 2729 CrossRef CAS.
- J. L. Shui, C. Chen and J. C. M. Li, Adv. Funct. Mater., 2011, 21, 3357 CrossRef CAS.
- C. X. Xu, L.Q. Wang, R. Y. Wang and Y. Ding, Adv. Mater., 2009, 21, 2165 CrossRef CAS.
- C. X. Xu, R. Y. Wang, M. W. Chen, Y. Zhang and Y. Ding, Phys. Chem. Chem. Phys., 2010, 12, 239 RSC.
- X. G. Wang, J. Frenzel, W. M. Wang, H. Ji, Z. Qi, Z. H. Zhang and G. Eggeler, J. Phys. Chem. C, 2011, 115, 4456 CAS.
- H. Ji, X. G. Wang, C. C. Zhao, C. Zhang, J. L. Xu and Z. H. Zhang, CrystEngComm, 2011, 13, 2617 RSC.
- L. L. Zhang, T. H. Lu, J. C. Bao, Y. W. Tang and C. Li, Electrochem. Commun., 2006, 8, 1625 CrossRef CAS.
- Z. X. Liang, T. S. Zhao, J. B. Xu and L. D. Zhu, Electrochim. Acta, 2009, 54, 2203 CrossRef CAS.
- Y. Zhu, Y. Y. Kang, Z. Q. Zou, Q. Zhou, J. W. Zheng, B. J. Xia and H. Yang, Electrochem. Commun., 2008, 10, 802 CrossRef CAS.
- F. L. Jia, K. W. Wong and R. X. Du, Electrochem. Commun., 2009, 11, 519 CrossRef CAS.
- F. L. Jia, K.W. Wong and L. Z. Zhang, J. Phys. Chem. C, 2009, 113, 7200 CAS.
- C. W. Xu, H. Wang, P. K. Shen and S. P. Jiang, Adv. Mater., 2007, 19, 4256 CrossRef CAS.
- Z. Yin, H. J. Zheng, D. Ma and X. H. Bao, J. Phys. Chem. C, 2009, 113, 1001 CAS.
- X. G. Wang, W. M. Wang, Z. Qi, C. C. Zhao, H. Ji and Z. H. Zhang, Electrochem. Commun., 2009, 11, 1896 CrossRef CAS.
- X. G. Wang, W. M. Wang, Z. Qi, C. C. Zhao, H. Ji and Z. H. Zhang, J. Power Sources, 2010, 195, 6740 CrossRef CAS.
- H. J. Qiu and F. X. Zou, ACS Appl. Mater. Interfaces, 2012, 4, 1404 CAS.
- C. X. Xu, Q. Li, Y. Q. Liu, J. P. Wang and H. R. Geng, Langmuir, 2012, 28, 1886 CrossRef CAS.
- H. J. Qiu and X. R. Huang, J. Mater. Chem., 2012, 22, 7602 RSC.
- H. J. Qiu, L. Li, Q. L. Lang, F. X. Zou and X. R. Huang, RSC Adv., 2012, 2, 3548 RSC.
- C. X. Xu, Y. Q. Liu, J. P. Wang, H. R. Geng and H. J. Qiu, J. Power Sources, 2012, 199, 124 CrossRef CAS.
- C. C. Zhao, Z. Qi, X. G. Wang and Z. H. Zhang, Corros. Sci., 2009, 51, 2120 CrossRef CAS.
- J. R. Mellor, N. J. Coville, S. H. Durbach and R. G. Copperthwaite, Appl. Catal., A, 1998, 171, 273 CrossRef CAS.
- K. Sieradzki, N. Dimitrov, D. Movrin, C. McCall, N. Vasiljevic and J. Erlebacher, J. Electrochem. Soc., 2002, 149, B370 CrossRef CAS.
- A. Dursun, D. V. Pugh and S.G. Corcoran, Electrochem. Solid-State Lett., 2003, 6, B32 CrossRef CAS.
- M. Grdeń, A. Piascik, Z. Koczorowski and A. Czerwiński, J. Electroanal. Chem., 2002, 532, 35 CrossRef.
- J. Snyder, P. Asanithi, A. B. Dalton and J. Erlebacher, Adv. Mater., 2008, 20, 4883 CrossRef CAS.
- H. Ji, J. Frenzel, Z. Qi, X. G. Wang, C. C. Zhao, Z. H. Zhang and G. Eggeler, CrystEngComm, 2010, 12, 4059 RSC.
- I. Beszeda, E. G. Gontier-Moya and D. L. Beke, Surf. Sci., 2003, 547, 229 CrossRef CAS.
- J. Erlebacher, J. Electrochem. Soc., 2004, 151, C614 CrossRef CAS.
- H. Kaiser, Corros. Sci., 1993, 34, 683 CrossRef CAS.
- J. T. Zhang, M. H. Huang, H. Y. Ma, F. Tian, W. Pan and S. H. Chen, Electrochem. Commun., 2007, 9, 1298 CrossRef CAS.
- L. H. Dall'Antonia, G. T. Filho and G. Jerkiewicz, J. Electroanal. Chem., 2001, 502, 72 CrossRef CAS.
- M. Grdeń, M. Łukaszewski, G. Jerkiewicz and A. Czerwiński, Electrochim. Acta, 2008, 53, 7583 CrossRef.
- S. Trasatti and O. A. Petrii, Pure Appl. Chem., 1991, 63, 711 CrossRef CAS.
- M. W. Xu, G. Y. Gao, W. J. Zhou, K. F. Zhang and H. L. Li, J. Power Sources, 2008, 175, 217 CrossRef CAS.
- P. Mani, R. Srivastava and P. Strasser, J. Phys. Chem. C, 2008, 112, 2770 CAS.
- P. Strasser, S. Koh, T. Anniyev, J. Greeley, K. More, C. F. Yu, Z. C. Liu, S. Kaya, D. Nordlund, H. Ogasawara, M. F. Toney and A. Nilsson, Nat. Chem., 2010, 2, 454 CrossRef CAS.
- A. U. Nilekar, Y. Xu, J. Zhang, M. B. Vukmirovic, K. Sasaki, R. R. Adzic and M. Mavrikakis, Top. Catal., 2007, 46, 276 CrossRef CAS.
- F. F. Onana and O. Savadogo, Electrochim. Acta, 2009, 54, 1769 CrossRef.
- V. Stamenkovic, B. S. Mun, K. J. J. Mayrhofer, P. N. Ross, N. M. Markovic, J. Rossmeisl, J. Greeley and J. K. Norskov, Angew. Chem., Int. Ed., 2006, 45, 2897 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |