Self-assembled nanostructures of Ag6[PV3Mo9O40] with N-donor ligands and their catalytic activity†
Bingfeng
Chen
ab,
Fengbo
Li
*a,
Zhijun
Huang
ab,
Tao
Lu
ab,
Yin
Yuan
ab,
Jialu
Yu
c and
Guoqing
Yuan
*a
aBeijing National Laboratory of Molecular Science, Laboratory of New Materials. Institute of Chemistry, Chinese Academy of Sciences, Beijing, P. R. China. E-mail: lifb@iccas.ac.cn; yuangq@iccas.ac.cn; Fax: (86)-10-62559373; Tel: (86)-10-62634920;
bGraduate University of Chinese Academy of Sciences, Beijing, 100049, P. R. China
cDepartment of Chemical Engineering, Tainjin University, Tainjin, 300072, P. R. China
First published on 18th October 2012
Abstract
Four types of nanostructures were obtained by self-assembly of Ag6[PV3Mo9O40] with N-donor ligands. Monodentate ligands lead to zero-dimensional nanostructures (nanoparticles). Linear didentate bridging ligands result in linear nanorods. Planar didentate chelating ligands develop two-dimensional nanostructures (nanosheets). Nonplanar didentate chelating ligands produce a transition morphology between nanorods and nanosheets. Kinetically driven (Utotal(x) < 0) self-assembly processes lead to structures with less defined shapes. Thermodynamically driven self-assembly (Utotal(x) is close to zero) produces ordered nanostructures. It is demonstrated that these nanocomposites are active catalysts for activating molecular oxygen under mild conditions.
Introduction
Self-assembly is defined as the spontaneous association of two or more molecules or ions to create a larger, aggregate species through the formation of reversible (generally supramolecular) interactions.1 The concepts of self-assembly are based on molecules, but components of any size can self-assemble in a permissive environment.2 With expanding contact of chemistry with biology and materials science, there are three important ranges of self-assembly component size: molecular, nanoscale (colloids, nanoparticles, nanorods, nanowires and related structures), and meso- to macroscopic. Self-assembly provides a simple and controllable strategy for fabrication of useful structures of nano- and macroscale components, which have great potential applications in microelectronics,3,4 photonics,5,6 near-field optics,7 and the emerging field of nanoscience.8 Solution-based self-assembly of inorganic nanoclusters can generate a variety of structures, including chains,9 sheets,10 vesicles,11 three-dimensional (3D) crystals12 or more complex 3D architectures.13 Such assembly is directed by the balance of attractive and repulsive forces.Polyoxometalates (POM) are polymeric oxoanions of early transition metals and can be formulated as [MmOy]p− (isopolyoxometalates) and [XxMmOy]q− (x < m) (heteropolyoxometalates), where M is the addenda atom and X is the heteroatom. When selecting certain constituent elements and counter-cations, they exhibit various chemical properties such as acidity, redox potentials, and solubility in many media. These properties stimulate broad research in their applications in catalysis, materials, and medicine.14 The spherical surface and the terminal bridging oxygen atoms of POM anions give a better opportunity to be used as inorganic building units for the aesthetic construction of supramolecular arrays.15
Supramolecular structures of giant polyoxometalate clusters have been reviewed by Müller et al.16 Supramolecular structures from anionic POM clusters and organic cations–coordination complex cations have been extensively explored.17 Supramolecular self-assembly features of polyoxometalate with transition metal complexes were investigated in the work.
Many polyoxometalates are effective catalysts for green oxidation using H2O2 or O2 as oxidants.18 However, most of these oxidations are homogeneous and problematic in recycling the catalysts and separating the products. In practical applications, there is a quest for the development of recoverable and recyclable heterogeneous oxidation catalysts. There are two strategies for heterogenizing: (1) solidification of polyoxometalates through the formation of insoluble solid ionic materials with selected countercations;19 (2) immobilization of polyoxometalates through adsorption, covalent linkage and ion-exchange.20 Polyoxometalates can be used as building blocks for many novel functional materials. This is a new horizon for the development of heterogeneous polyoxometalate-based oxidation catalysts.21 Self-assembly is an efficient and low-cost method for exploring novel nanocatalysts based on POM building units.
Phosphovanadomolybdate Keggin-type polyoxometalates (H3+xPVxMo12−xO40 (x = 1–6)) have a redox potential of about 0.7 V vs. SHE, which is lower than that of O2. These compounds can activate molecular oxygen for homogeneous oxidation under mild conditions.22 Silver salt of phosphovanadomolybdate shows high thermal stability (>800 °C) and has great potential in acting as a heterogeneous catalyst for controllable oxidation with molecular oxygen.23,24 In this work, Ag6[PV3Mo9O40] was prepared with the same procedure as its analogy (Ag5[PV2Mo10O40]).23 Polymeric coordinate materials were formed through self-assembly with nitrogen ligands in CH3CN solution. Different N-ligands lead to different self-assembled nanostructures, and several of them exhibit high activity in catalyzing C–H direct oxidation with 1.0 atm oxygen.
Experimental
Materials
All chemicals were purchased from Sinopharm Chemical Reagent Co. Ltd. Acetonitrile was distilled and purified before using. All the reagents were used as received.Results and discussion
Self-assembled nanostructures of Ag6[PV3Mo9O40] with N-donor ligands
Monovalent silver (Ag+) exhibits a high coordinative flexibility in coordination numbers between 2 and 6 and in typical closed-shell ion polyhedra.24 Strong N-donor ligands coordinate Ag+ linearly in cationic complexes, with trigonal or tetrahedral environments, depending on the strength of the competing ligands and stoichiometry.25,26 For Ag6[PV3Mo9O40], Ag+ is bonded to the phosphovanadomolybdate polyanion with strong static electronic interaction. The Ag+ is polarizable and has a high tendency to coordinate soft Pearson bases (aromatic N-donor ligands) with covalent bonding contributions. When introducing N-donor ligands into a CH3CN solution of Ag6[PV3Mo9O40] (1.0 × 10−3 M), the self-assembly of the POM cluster with N-donor ligands occurs through [AgI←N] coordinate interaction. In our experiments, twelve aromatic N-donor ligands were attempted. Four typical self-assembled nanostructures were obtained (as shown in Fig. 1).![(a) Zero-dimensional nanostructures, nanoparticles (1a), from the self-assembly of Ag6[PV3Mo9O40] with mononitrogen donor benzoimidazole. (b) One-dimensional nanostructures, nanorods (1b), from the self-assembly of Ag6[PV3Mo9O40] with 4,4′-dipyridine. (c) Self-aggregate balance of Ag6[PV3Mo9O40] clusters in CH3CN solution. (d) Self-assembled nanostructures (1d) from Ag6[PV3Mo9O40] and 2,2′-dipyridine. (e) Two-dimensional nanostructures, nanosheets (1e), from the self-assembly of Ag6[PV3Mo9O40] with 1,10-phenanthroline.](/image/article/2012/RA/c2ra21858e/c2ra21858e-f1.gif) | ||
| Fig. 1 (a) Zero-dimensional nanostructures, nanoparticles (1a), from the self-assembly of Ag6[PV3Mo9O40] with mononitrogen donor benzoimidazole. (b) One-dimensional nanostructures, nanorods (1b), from the self-assembly of Ag6[PV3Mo9O40] with 4,4′-dipyridine. (c) Self-aggregate balance of Ag6[PV3Mo9O40] clusters in CH3CN solution. (d) Self-assembled nanostructures (1d) from Ag6[PV3Mo9O40] and 2,2′-dipyridine. (e) Two-dimensional nanostructures, nanosheets (1e), from the self-assembly of Ag6[PV3Mo9O40] with 1,10-phenanthroline. | ||
It is a generally accepted fact that soluble inorganic ions should distribute homogeneously in dilute solutions. But soluble POM macroanions in a polar solvent usually do not exist as single ions. The Tyndall effect was observed from the POM solutions.27 The Tyndall effect is defined as the visible light scattering from suspended particles of colloidal size. In the solution of Ag6[PV3Mo9O40], the balance of self-aggregate of primary units is the existing pattern of POM clusters (Fig. 1c).
Zero-dimensional nanostructures, nanoparticles (1a), result from the self-assembly of Ag6[PV3Mo9O40] with mononitrogen donor benzoimidazole (Fig. 1a). Nanoparticles are dispersed well and their average size is about 10 nm. One-dimensional nanostructures, nanorods (1b), were prepared by the self-assembly of Ag6[PV3Mo9O40] with 4,4′-dipyridine (Fig. 1b). Two-dimensional nanostructures, nanosheets (1e), were prepared by introducing 1,10-phenanthroline into Ag6[PV3Mo9O40] CH3CN solution (Fig. 1e). Fig. 1d shows nanostructures (1d) between nanorods and nanosheets, which were prepared through the procedure involving 2, 2′-dipyridine. It is revealed that the morphology of self-assembled nanostructures has a subtle relation to the molecular structure of N-donor ligands. Monodentate ligands lead to zero-dimensional nanostructures, nanoparticles. Linear didentate bridging ligands result in linear nanorods. Planar didentate chelating ligands develop two- dimensional nanostructures, nanosheets.
Nonplanar didentate chelating ligands produce a transition morphology between nanorods and nanosheets. Sub-structures of self-assembled nanostructures were further characterized by TEM with higher magnification. Fig. 2b–d shows closer views of the sub-structures of nanorods (1b). It is revealed that nanorods are built up by arranging sub-nanostructures, nanoclusters with size of about 1.0 nm, which meet the scale of a Ag6[PV3Mo9O40] cluster.23,24 The nanoparticles (10 nm) in Fig. 1a are self-assembled structures of Ag6[PV3Mo9O40] with mononitrogen donor benzoimidazole. The silver 3d5/2 XPS peak of Ag6[PV3Mo9O40] has a binding energy of 368.2 eV, which is indexed to a AgI ion chelated to oxygen.28 In nanorods from 4,4′-dipyridine and Ag6[PV3Mo9O40], the binding energy of Ag 3d5/2 shows a marked shift and the peak is broadened (Fig. 3). The [AgI←N] coordinate interaction can increase the electron density of the AgI ion and lower the binding energy of Ag 3d5/2. The involvement of the N-donor complicates the chemical state of the AgI ion and expands the half peak width of Ag 3d5/2.
![(a) Nanorods from the self-assembly of Ag6[PV3Mo9O40] with 4,4′-dipyridine. (b), (c), (d) TEM images of sub-structures of nanorods.](/image/article/2012/RA/c2ra21858e/c2ra21858e-f2.gif) | ||
| Fig. 2 (a) Nanorods from the self-assembly of Ag6[PV3Mo9O40] with 4,4′-dipyridine. (b), (c), (d) TEM images of sub-structures of nanorods. | ||
![Ag 3d5/2, Ag 3d3/2 XPS peaks of Ag6[PV3Mo9O40] and nanorods (1b).](/image/article/2012/RA/c2ra21858e/c2ra21858e-f3.gif)
The correlation between nanoscale nanostructures and self-assembly conditions in solution
Fig. 1c illustrates the self-assembly of Ag6[PV3Mo9O40] clusters with N-ligands in CH3CN solution. Assembly is controlled by the balance of attractive and repulsive forces.29 Suppose that Utotal(x) is the total net potential of the entire self-assembly process with AgPOM clusters. The Utotal(x) can be formulated as the net total of all of the attractive and repulsive potentials involved in each step of the self-assembly:30| Utotal(x) = fP|UA,P(x) + UR,P(x)| + fS|UA,S(x) + UR,S(x)| + fH|UA,H(x) + UR,H(x)| + ··· + Uext(x) |
with
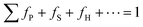 | (1) |
U A,P(x) is the attractive potential of the self-assembly of primary building units and UR,P(x) represents the repulsive potentials. UA,S(x) and UR,S(x) are for the self-assembly of secondary building units, and UA,H(x) and UR,H(x) are for the self-assembly of higher-order building units. fP, fS, and fH are the fraction coefficients of each self-assembly step to the total net potential. Uext(x) is the potential contribution from the external forces.
For the self-assembly of Ag6[PV3Mo9O40] clusters with N-ligands in CH3CN solution, Uext(x) is zero. When Ag6[PV3Mo9O40] is dissolved in CH3CN, Utotal(x) is equal to zero for the dissolution balance. The presence of strong chelating N-ligands for silver ion can change the chemical environment around the Ag6[PV3Mo9O40] clusters. This affects the balance of attractive and repulsive forces, which govern the self-assembly processes.
There are three possibilities:31 when Utotal(x) is positive for the new system, the self-assembly is not possible in most cases. When Utotal(x) is negative, the self-assembly is kinetically driven. The self-assembly occurs until most of the primary units are exhausted. The resulting structures show indefinite sizes and poor physical shapes. The higher the value of |Utotal(x)|, the faster the self-aggregates form. When Utotal(x) is equal to or close to zero, the self-assembly is thermodynamically driven. The self-assembly steps are in or close to the equilibrium with the self-aggregates. The resulting structures have finite sizes and well-defined shapes.
In our experiments, the self-assembly of Ag6[PV3Mo9O40] with N-donor ligands was carried out in diluted CH3CN solution. Ag6[PV3Mo9O40] was dissolved in extra pure and dry CH3CN (100 ml, 1.0 × 10−3 M). CH3CN solution of an N-donor ligand (10 ml, 0.1 M) was added by automatic microinjector with gentle stirring. The rate of injection of an N-ligand solution is a key parameter governing the final self-assembled nanostructures. In the self-assembly of Ag6[PV3Mo9O40] with benzoimidazole, the structures shown in Fig. 4a were obtained by the injection of 10 ml CH3CN solution containing 1.0 mmol benzoimidazole in 5.0 min. The structure in Fig. 4b was synthesized in 1.0 h and the injection time of Fig. 4c was 3.0 h. Well-defined nanoparticles were achieved through 6.0 h injection of 10 ml CH3CN solution containing 1 mmol benzoimidazole (Fig. 4d). It is clearly revealed that the fast introduction of N-ligands leads to structures with indefinite sizes and poor physical shapes. The controlled and gradual introduction of N-ligands results in well-defined nanostructures. The same phenomena were observed in the self-assembly processes of Ag6 [PV3Mo9O40] with 4,4′-dipyridine (Fig. 5) and 1,10-phenanthroline (Fig. 6).
![TEM images of self-assembled structures of Ag6[PV3Mo9O40] clusters with benzoimidazole in CH3CN solution. Self-assembly time: (a) 5.0 min; (b) 1.0 h; (c) 3.0 h; (d) 6.0 h.](/image/article/2012/RA/c2ra21858e/c2ra21858e-f4.gif) | ||
| Fig. 4 TEM images of self-assembled structures of Ag6[PV3Mo9O40] clusters with benzoimidazole in CH3CN solution. Self-assembly time: (a) 5.0 min; (b) 1.0 h; (c) 3.0 h; (d) 6.0 h. | ||
![Images of self-assembled structures of Ag6[PV3Mo9O40] clusters with 4,4′-dipyridine in CH3CN solution. Self-assembly time: (a) 5.0 min; (b) 1.0 h; (c) 3.0 h; (d) 6.0 h.](/image/article/2012/RA/c2ra21858e/c2ra21858e-f5.gif) | ||
| Fig. 5 Images of self-assembled structures of Ag6[PV3Mo9O40] clusters with 4,4′-dipyridine in CH3CN solution. Self-assembly time: (a) 5.0 min; (b) 1.0 h; (c) 3.0 h; (d) 6.0 h. | ||
![TEM images of self-assembled structures of Ag6[PV3Mo9O40] clusters with 1,10-phenanthroline in CH3CN solution. Self-assembly time: (a) 5.0 min; (b) 1.0 h; (c) 3.0 h; (d) 6.0 h.](/image/article/2012/RA/c2ra21858e/c2ra21858e-f6.gif) | ||
| Fig. 6 TEM images of self-assembled structures of Ag6[PV3Mo9O40] clusters with 1,10-phenanthroline in CH3CN solution. Self-assembly time: (a) 5.0 min; (b) 1.0 h; (c) 3.0 h; (d) 6.0 h. | ||
If the disturbance of N-ligands cannot be buffered (with a high rate of injection of N-ligands), the self-assembly is kinetically driven (Utotal(x) < 0). Less-defined structures were obtained (Fig. 4a, Fig. 4b, Fig. 5a, Fig. 5b, Fig. 6a, and Fig. 6b). When the introduction of N-ligands was in a controlled and gradual way, the self-assembly is thermodynamically driven (Utotal(x) is close to zero). Well-defined nanostructures were achieved (Fig. 4d, Fig. 5d, and Fig. 6 d).
As discussed above, self-assembled nanostructures have subtle relations with the molecular structure of N-ligands. Chemical heterogeneity around Ag6[PV3Mo9O40] clusters induces site-specific interactions with N-ligands in the solution-based self-assembly.32 Coordinative flexibility of silver ions and N-ligands generates a variety of structures. Monodentate ligands lead to zero-dimensional nanostructures (nanoparticles, Fig. 4d). Linear didentate bridging ligands result in linear nanorods (Fig. 5d). Planar didentate chelating ligands develop two-dimensional nanostructures (nanosheets, Fig. 6d). Nonplanar didentate chelating ligands produce transition morphology between nanorods and nanosheets (Fig. 1c).
Catalytic activity
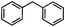
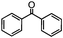
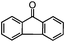
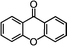
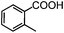
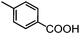
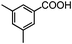
Self-assembly has been recognized as a powerful strategy for manipulating the structure and properties of ensembles of inorganic nanoparticles.33 N-ligand directed ensembles of Ag6[PV3Mo9O40] nanoclusters exhibit collective properties, which are different from those of individual polyoxometalate cluster and bulk materials. H3+xPVxMo12−xO40 (x = 1–6) phosphovanadomolybdate Keggin-type polyoxometalates are active catalysts for oxygen activation.22 Silver salt of phosphovanadomolybdate shows a high thermal stability (>800 °C) and has great potential in acting as a heterogeneous catalyst for controllable oxidation with molecular oxygen.23,24 The catalytic activity of self-assembled nanostructures was tested for the direct oxidation of the C–H bond adjacent to aromatic cycle. The analogous reactions over homogeneous polyoxometalate systems have been explored through well-designed methods, as listed in Table S1, ESI.† In our experimental observation, these as-synthesized nanostructures have improved catalytic activity.Oxidation of diphenylmethane to benzophenone with oxygen was selected as a model reaction. The catalyst 1b shows the highest activity in solvent of benzonitrile at 100 °C (as shown in Fig. S1, ESI†). Table 1 lists the experimental results of various heterogeneous silver catalysts and control tests without silver polyoxometalates. It is demonstrated that 1a, 1b, 1d, and 1e are efficient catalysts for activation of molecular oxygen. The conversions of the substrate under 1b and 1e are near 100%. The substrates for catalytic oxidation are expanded, as shown in Table 2. Nanocatalyst 1b show highly catalytic activity for diphenylmethane, fluorene, and 9H-xanthene to produce corresponding ketones. Oxidation of triphenylmethane leads to a complex mixture and shows only 36% selectivity to triphenylmethanol. Oxyfunctionalization of alkyl aromatics is an important industrial process for producing intermediates for plastic, alkyd and polyester resins, dyes, and numerous fine chemicals. General catalytic processes are carried out over V2O5 catalysts at 350–550 °C with excess air. Homogeneous liquid-phase oxidation (Rhône-Progil process) can proceed at a low temperature of 150–200 °C, but requires high radical catalyst concentration with cocatalysts containing bromine. In our experimental observation, heterogeneous nanocatalyst 1b, shows medium catalytic activity for selective oxidation of 1,2,3,4-tetrahydronaphthalene, 1,2,4,5-tetramethylbenzene, and 1,3,5-trimethylbenzene at 100 °C with 1.0 atm oxygen. Unexpectedly, phthalic anhydride is formed from oxidation of o-xylene under such mild reaction conditions. Nanocatalysts from the self-assembly of twelve N-donor ligands and Ag6[PV3Mo9O40] were investigated and the results were listed in Table S2.† There are two possible reaction mechanisms: (1) the direct activation of molecular oxygen by the catalyst, (2) a Fenton-type radical chain mechanism. N-ligand directed ensembles of Ag6[PV3Mo9O40] nanoclusters exhibit collective properties, which include these two catalytic properties.
| Catalyst | Conversion/selectivity (%) |
|---|---|
| a Reaction conditions: 2.0 mmol diphenylmethane, 1.0 ml PhCN, 100 °C, oxygen balloon, 50 mg catalyst, reaction time 10 h. | |
| Blank | <5.0 |
| AgCl | 11.3/78.3 |
| Ag2MoO4 | 13.4/72.2 |
| AgVO3 | 21.4/83.5 |
| Ag6[PV3Mo9O40] | 48.6/90.1 |
| Na6PV3Mo9O40 | 29.7/87.5 |
| H6PV3Mo9O40 | 34.1/91.2 |
| Benzoimidazole | <5.0 |
| 1a | 98.2/95.2 |
| 4,4′-Dipyridine | <5.0 |
| 1b | >99.9/99.2 |
| 2,2′-Dipyridine | <5.0 |
| 1d | 97.8/94.3 |
| 1,10-Phenanthroline | 6.7 |
| 1e | >99.9/98.7 |
 99.2
99.2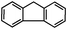
 99.0
99.0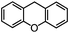
 99.0
99.0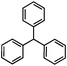
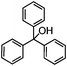 36
36
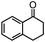 95.4
95.4
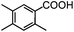 89.6
89.6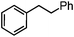
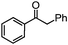 36
36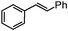 47
47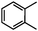
 75
75 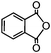 25
25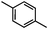
 90.4
90.4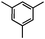
 86.7
86.7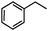
 62.3
62.3Phthalic anhydride (PA) is an important chemical for the manufacture of phthalate plasticizers, alkyd and polyester resins, and phthalocyanine dyes. The word capacity for PA was about 4.5 × 106 t/a in 2005.34 BASF has developed commercial gas-phase oxidation of o-xylene to PA.35 The process is carried out at 375–410 °C over V2O5–TiO2 catalysts in multitube reactors with about 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tubes. The Air/feed ratio is 18:1 and the contact time of feed with the catalyst is <0.5 s. The oxidation of o-xylene to PA by 1b was further investigated in a one-batch nonsolvent test (Fig. 7). The conversion of o-xylene increases with the pressure of oxygen in the sealed autoclave and the selectivity to PA reaches 40% at 100 °C under 3.0 MPa oxygen.
000 tubes. The Air/feed ratio is 18:1 and the contact time of feed with the catalyst is <0.5 s. The oxidation of o-xylene to PA by 1b was further investigated in a one-batch nonsolvent test (Fig. 7). The conversion of o-xylene increases with the pressure of oxygen in the sealed autoclave and the selectivity to PA reaches 40% at 100 °C under 3.0 MPa oxygen.
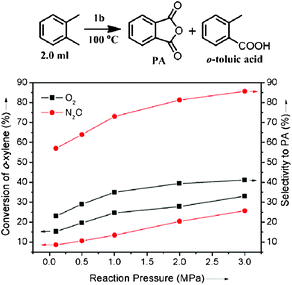 | ||
| Fig. 7 Nonsolvent oxidation of o-xylene to phthalic anhydride catalyzed by 1b with O2 and N2O. Reaction conditions: 2.0 ml o-xylene, 100 °C, O2 or N2O, 50 mg catalyst, 25 ml autoclave with a magnetic stir, reaction time 10 h. | ||
Under the same reaction conditions, there are no oxidation reactions over the V2O5–TiO2 catalyst.36 Heterogeneous Co and Mn catalysts for liquid phase o-xylene oxidation show the selectivity (5 mol %) to PA at the o-xylene conversion of 20 mol% under 172 °C.37 N2O was found to be the active oxidant for converting o-xylene to PA over nanocatalyst 1b. PA is the main product and its selectivity reaches 85% at the conversion of 25% at 100 °C under 3.0 MPa N2O (Fig. 7). N2O is a safer oxidant than oxygen. Higher reaction pressure can be attempted to increase the conversion of o-xylene. This shows a possibility of developing a mild oxidation process of o-xylene to PA based on nanocatalyst 1b. The mild oxidation process can abate the byproducts and avoid the danger of explosion and CO2 from total oxidation at a high reaction temperature. However, the converting efficiency must be improved. There are two solutions: (1) improving the catalytic activity of the catalysts, (2) improving feed recycling and contact with catalyst in the reactors, like the multitube reactor of the BASF process. Further investigation on this topic is under way.
The recyclability of these nanocomposites was investigated by a five-run test of the catalytic oxidation of diphenylmethane over 1b. After each test run, the catalyst was separated by filtration and washed three times with acetone, then dried for the next run. The catalytic activity was denoted by TOF. Fig. 8a shows the results of the five-run test. After the fifth test, there is a nearly 25% decrease in the catalytic activity. Fig. 8b is the TEM image of the used catalyst. The one-dimensional nanostructure (nanorod), can be clearly discriminated. The nanorod is built up by arranging sub-nanostructures (Ag6[PV3Mo9O40] clusters), as observed in the TEM image of the fresh catalyst (1b) (Fig. 2b). This proves that the nanostructure of the used catalyst remains constant. During the catalytic process, the leaching out of some active species may take place. The decomposition of edge sites is the main reason that causes leaching of the catalyst. The concentration of silver ions in the reaction solvent can be used as a quantitative index to the degree of catalytic stability. If a high concentration of silver ions were detected after the reaction, serious decomposition of the catalyst might happen. Fig. 8c shows the silver ion concentrations in the reaction solvent after each test run. After the first test run, the leaching silver concentration is 124 ppb. In the following test runs, the value is in the range 20 to 40 ppb. The relatively high concentration of leaching silver in the first run is attributed to loosely chelated Ag6[PV3Mo9O40] clusters in the surface edge defects. However, the leaching amount on such a negligible scale shows no marked influence on the catalytic behavior. This has been confirmed by Fig. 8a and Fig. 8b. In summary, self-assembled nanostructures of Ag6[PV3Mo9O40] with N-donor ligands can tolerate the operation conditions of the liquid-phase oxidation process. They can be recycled efficiently.
 | ||
| Fig. 8 a) Five-run test of recyclability of 1b. Reaction conditions: 2.0 mmol diphenylmethane, 1.0 ml PhCN, 100 °C, oxygen balloon, 50 mg catalyst, reaction time 10 h. (b) TEM image of the used catalyst. (c) The silver ion concentrations in the reaction solvent after each test run. | ||
Conclusion
In summary, four types of nanostructures were obtained by self-assembly of Ag6[PV3Mo9O40] with N-donor ligands. Monodentate ligands lead to zero-dimensional nanostructures, nanoparticles. Linear didentate bridging ligands result in linear nanorods. Planar didentate chelating ligands develop two-dimensional nanostructures, nanosheets. Nonplanar didentate chelating ligands produce a transition morphology between nanorods and nanosheets. Kinetically driven self-assembly (Utotal(x) < 0) leads to less-defined structures. When the introduction of N-ligands took place in a controlled and gradual way, the self-assembly is thermodynamically driven (Utotal(x) is close to zero). Well-defined nanostructures were achieved.It is demonstrated that these nanocomposites are active catalysts for activating molecular oxygen under mild conditions. In oxyfunctionalization of alkyl aromatics, nanorods (1b) show highly catalytic activity for diphenylmethane, fluorene, and 9H-xanthene to produce the corresponding ketones, and medium catalytic activity for selective oxidation of 1,2,3,4-tetrahydronaphthalene, 1,2,4,5-tetramethylbenzene, and 1,3,5-trimethylbenzene at 100 °C with 1.0 atm oxygen. Phthalic anhydride is formed through 1b catalyzed oxidation of o-xylene under such mild reaction conditions. Under nonsolvent reaction with 3.0 MPa O2 in a sealed autoclave, the selectivity to PA increases to 40% at 33.1% conversion. Under the same conditions, N2O shows 85% selectivity to PA at 25.3% conversion of o-xylene.
Acknowledgements
This work was financially supported by the National Natural Sciences Foundation of China (no. 21101161, 21174148).References
- G. F. Swiegers, Self-assembly: Terminology in Encyclopedia of Supramolecular Chemistry, ed. J. W. Steed and J. L. Atwood, Marcel Dekker, New York, 2004, pp. 1263–1269 Search PubMed.
- L. Isaacs, D. N. Chin, N. Bowden, Y. Xia and G. M. Whitesides, in Supramolecular Technology, ed. D. N. Reinhoudt, Wiley, New York, 1999, pp. 1–46 Search PubMed.
- H. Sirringhaus, T. Kawase, R. H. Friend, T. Shimoda, M. Inbasekaran, W. Wu and E. P. Woo, Science, 2000, 290, 2123–2126 CrossRef CAS.
- J. A. Rogers, Z. Bao, K. Baldwin, A. Dodabalapur, B. Crone, V. R. Raju, V. Kuck, H. Katz, K. Amundson, J. Ewing and P. Drzaic, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 4835–4840 CrossRef CAS.
- S. A. Jenekhe and L. X. Chen, Science, 1999, 283, 372–375 CrossRef CAS.
- Y. Xia, B. Gates, Y. Yin and Y. Lu, Adv. Mater., 2000, 12, 693–713 CrossRef CAS.
- M. H.Wu and G. M. Whitesides, Appl. Phys. Lett., 2001, 78, 2273–2275 CrossRef CAS.
- (a) C. M. Lieber, Sci. Am., 2001, 285, 58–64 CrossRef CAS; (b) V. I. Klimov, A. A. Mikhailovski, S. Xu, A. Malko, J. A. Hollingsworth, C. A. Leatherdale, H. J. Eisler and M. G. Bawendi, Science, 2000, 290, 314–317 CrossRef CAS; (c) S. H. Sun, C. B. Murray, D. Weller, L. Folks and A. Moser, Science, 2000, 287, 1989–1992 CrossRef CAS.
- (a) G. A. DeVries, M. Brunnbauer, Y. Hu, A. M. Jackson, B. Long, B. T. Neltner, O. Uzun, B. H. Wunsch and F. Stellacci, Science, 2007, 315, 358–361 CrossRef CAS; (b) Z. H. Nie, D. Fava, E. Kumacheva, S. Zou, G. C. Walker and M. Rubinstein, Nat. Mater., 2007, 6, 609–614 CrossRef CAS; (c) Y. J. Kang, K. J. Erickson and T. A. Taton, J. Am. Chem. Soc., 2005, 127, 13800–13801 CrossRef CAS; (d) K. K. Caswell, J. N. Wilson, U. H. F. Bunz and C. J. Murphy, J. Am. Chem. Soc., 2003, 125, 13914–13915 CrossRef CAS.
- (a) Z. Y. Tang, Z. L. Zhang, Y. Wang, S. C. Glotzer and N. A. Kotov, Science, 2006, 314, 274–278 CrossRef CAS; (b) N. N. Zhao, K. Liu, J. Greener, Z. H. Nie and E. Kumacheva, Nano Lett., 2008, 9, 3077–3081 Search PubMed.
- S. Park, J. H. Lim, S. W. Chung and C. A. Mirkin, Science, 2004, 303, 348–351 CrossRef CAS.
- (a) A. M. Kalsin, M. Fialkowski, M. Paszewski, S. K. Smoukov, K. J. M. Bishop and B. A. Grzybowski, Science, 2006, 312, 420–424 CrossRef CAS; (b) D. Nykypanchuk, M. M. Maye, D. van der Lelie and O. Gang, Nature, 2008, 451, 549–552 CrossRef CAS; (c) S. Y. Park, A. K. R. Lytton-Jean, B. Lee, S. Weigand, G. C. Schatz and C. A. Mirkin, Nature, 2008, 451, 553–556 CrossRef CAS; (d) C. R. Iacovella and S. C. Glotzer, Nano Lett., 2009, 9, 1206–1211 CrossRef CAS.
- J. Sharma, R. Chhabra, A. Cheng, J. Brownell, Y. Liu and H. Yan, Science, 2009, 323, 112–116 CrossRef CAS.
- (a) C. L. Hill, Chem. Rev., 1998, 98, 1–2 CrossRef CAS; (b) M. T. Pope, Heteropoly and Isopoly Oxometalates, Springer-Verlag, Berlin, 1983 Search PubMed; (c) M. T. Pope and A. Müller, Polyoxometalate Chemistry From Topology via Self-Assembly to Applications, Kluwer Academic Publishers, Dordrecht, 2001 Search PubMed; (d) T. Yamase and M. T. Pope, Polyoxometalate Chemistry for Nano-Composite Design, Kluwer Academic/Plenum Publishers, New York, 2002 Search PubMed; (e) M. T. Pope, Compr. Coord. Chem. II, 2004, 4, 635–678 Search PubMed; (f) P. Mialane, A. Dolbecq and F. Secheresse, Chem. Commun., 2006, 3477–3485 RSC; (g) D. L. Long, E. Burkholder and L. Cronin, Chem. Soc. Rev., 2007, 36, 105–121 RSC; (h) S. Uchida and N. Mizuno, Coord. Chem. Rev., 2007, 251, 2537–2546 CrossRef CAS; (i) C. L. Hill and C. M. Prosser- McCartha, Coord. Chem. Rev., 1995, 143, 407–455 CrossRef CAS; (j) T. Okuhara, N. Mizuno and M. Misono, Adv. Catal., 1996, 41, 113–252 CAS; (k) R. Neumann, Prog. Inorg. Chem., 1998, 47, 317–370 CAS; (l) N. Mizuno and M. Misono, Chem. Rev., 1998, 98, 199–218 CrossRef CAS; (m) I. V. Kozhevnikov, Chem. Rev., 1998, 98, 171–198 CrossRef CAS; (n) I. V. Kozhevnikov, Catalysis by Polyoxometalates, John Wiley & Sons, Chichester, 2002 Search PubMed; (o) N. Mizuno, K. Yamaguchi and K. Kamata, Coord. Chem. Rev., 2005, 249, 1944–1956 CrossRef CAS; (p) C. L. Hill, Compr. Coord. Chem. II, 2004, 4, 679–759 Search PubMed; (q) N. Mizuno, K. Kamata and K. Yamaguchi, in Surface and Nanomolecular Catalysis, ed. R. Richards, Taylor and Francis Group, New York, 2006 Search PubMed.
- (a) E. Coronado and J. Gomez-Garcia, Chem. Rev., 1998, 98, 273–296 CrossRef CAS; (b) C. L. Hill, Chem. Rev., 1998, 98, 1–2 CrossRef CAS; (c) J. T. Rhuie, C. L. Hill, D. A. Judd and R. F. Schinazi, Chem. Rev., 1998, 98, 327–358 CrossRef CAS.
- (a) A. Müller and C. Serain, Acc. Chem. Res., 2000, 33, 2–10 CrossRef CAS; (b) A. Müller, F. Peters, M. T. Pope and D. Gatteschi, Chem. Rev., 1998, 98, 239–272 CrossRef.
- (a) D. L. Long, E. Burkholder and L. Cronin, Chem. Soc. Rev., 2007, 36, 105–121 RSC; (b) A. Müller, P. Kögerler and A. W. M. Dress, Coord. Chem. Rev., 2001, 222, 193–218 CrossRef CAS.
- (a) R. Neumann and M. Dahan, Nature, 1997, 388, 353–355 CrossRef CAS; (b) I. A. Weinstock, E. M. G. Barbuzzi, M. W. Wemple, J. J. Cowan, R. S. Reiner, D. M. Sonnen, R. A. Heintz, J. S. Bond and C. L. Hill, Nature, 2001, 414, 191–195 CrossRef CAS; (c) K. Kamata, K. Yonehara, Y. Nakagawa, K. Uehara and N. Mizuno, Nat. Chem., 2010, 2, 478–483 CrossRef CAS; (d) K. Kamata, K. Yonehara, Y. Sumida, K. Yamaguchi, S. Hikichi and N. Mizuno, Science, 2003, 300, 964–966 CrossRef CAS; (e) Q. Yin, J. M. Tan, C. Besson, Y. V. Geletii, D. G. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle and C. L. Hill, Science, 2010, 328, 342–345 CrossRef CAS.
- (a) P. Nagaraju, N. Pasha, P. S. S. Prasad and N. Lingaiah, Green Chem., 2007, 9, 1126–1129 RSC; (b) Z. W. Xi, N. Zhou, Y. Sun and K. L. Li, Science, 2001, 292, 1139 CrossRef CAS; (c) V. K. Dloumaev and R. M. Bullock, Nature, 2003, 424, 530–532 CrossRef; (d) M. Clemente-León, C. Mingotaud, B. Agricole, C. J. Gómez-Garcia, E. Coronado and P. Delhaès, Angew. Chem., Int. Ed. Engl., 1997, 36, 1114–1116 CrossRef CAS; (e) M. Clemente-León, E. Coronado, P. Delhaes, C. J. Gómez-García and C. Mingotaud, Adv. Mater., 2001, 13, 574–577 CrossRef CAS; (f) M. V. Vasylyev and R. Neumann, J. Am. Chem. Soc., 2004, 126, 884–890 CrossRef CAS; (g) Y. M. A. Yamada, M. Ichinohe, H. Takahashi and S. Ikegami, Org. Lett., 2001, 3, 1837–1840 CrossRef CAS; (h) D. E. Bergbreiter, Chem. Rev., 2002, 102, 3345–3384 CrossRef CAS; (i) H. Hamamoto, Y. Suzuki, Y. M. A. Yamada, H. Tabata, H. Takahashi and S. Ikegami, Angew. Chem., Int. Ed., 2005, 44, 4536–4538 CrossRef CAS; (j) A. Haimov, H. Cohen and R. Neumann, J. Am. Chem. Soc., 2004, 126, 11762–11763 CrossRef CAS; (k) A. Haimov and R. Neumann, J. Am. Chem. Soc., 2006, 128, 15697–15700 CrossRef CAS.
- (a) L. Xu, E. Boring and C. L. Hill, J. Catal., 2000, 195, 394–405 Search PubMed; (b) R. D. Gall, C. L. Hill and J. E. Walker, J. Catal., 1996, 159, 473–478 CrossRef CAS; (c) A. N. Kharat, P. Pendleton, A. Badalyan, M. Abedini and M. M. Amini, J. Mol. Catal. A: Chem., 2001, 175, 277–283 CrossRef CAS; (d) R. Neumann, A. M. Khenkin and I. Vigdergauz, Chem.–Eur. J., 2000, 6, 875–882 CrossRef; (e) N. M. Okun, T. M. Anderson and C. L. Hill, J. Am. Chem. Soc., 2003, 125, 3194–3195 CrossRef CAS; (f) I. Bar-Nahum, A. M. Khenkin and R. Neumann, J. Am. Chem. Soc., 2004, 126, 10236–10237 CrossRef CAS; (g) K. Yamaguchi, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2000, 122, 7144–7145 CrossRef CAS; (h) K. Mori, K. Yamaguchi, T. Hara, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2002, 124, 11572–11573 CrossRef CAS; (i) K. Mori, T. Hara, T. Mizugaki, K. Ebitan and K. Kaneda, J. Am. Chem. Soc., 2003, 125, 11460–11461 CrossRef CAS; (j) K. Mori, T. Hara, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2004, 126, 10657–10666 CrossRef CAS; (k) G. Maayan and R. Neumann, Chem. Commun., 2005, 4595–4597 RSC; (l) A. Haimov and R. Neumann, Chem. Commun., 2002, 876–877 RSC; (m) R. Neumann and M. Cohen, Angew. Chem., Int. Ed. Engl., 1997, 36, 1738–1740 CrossRef CAS; (n) A. P. Wight and M. E. Davis, Chem. Rev., 2002, 102, 3589–3614 CrossRef CAS; (o) K. Yamaguchi, C. Yoshida, S. Uchida and N. Mizuno, J. Am. Chem. Soc., 2005, 127, 530–531 CrossRef; (p) T. Kwon, G. A. Tsigdinos and T. J. Pinnavaia, J. Am. Chem. Soc., 1988, 110, 3653–3654 CrossRef CAS; (q) B. Sels, D. De Vos, M. Buntinx, F. Pierard, A. Kirsch-De Mesmaeker and P. Jacobs, Nature, 1999, 400, 855–857 CrossRef CAS; (r) B. F. Sels, D. E. de Vos and P. A. Jacobs, J. Am. Chem. Soc., 2001, 123, 8350–8359 CrossRef CAS; (s) B. F. Sels, D. E. de Vos and P. A. Jacobs, Angew. Chem., Int. Ed., 2005, 44, 310–313 CrossRef CAS.
- (a) W. Bu, S. Uchida and N. Mizuno, Angew. Chem., Int. Ed., 2009, 48, 8281–8284 CrossRef CAS; (b) D. Long, R. Tsunashima and L. Cronin, Angew. Chem., Int. Ed., 2010, 49, 1736–1758 CrossRef CAS; (c) H. Li, H. Sun, W. Qi, M. Xu and L. Wu, Angew. Chem., Int. Ed., 2007, 46, 1300–1303 CrossRef CAS; (d) H. Zeng, G. R. Newkome and C. L. Hill, Angew. Chem., Int. Ed., 2000, 39, 1771–1774 CrossRef; (e) C. P. Pradeep, D. L. Long, G. N. Newton, Y. F. Song and L. Cronin, Angew. Chem., Int. Ed., 2008, 47, 4388–4391 CrossRef CAS; (f) C. Streb, R. Tsunashima, D. A. MacLaren, T. McGlone, T. Akutagawa, T. Nakamura, A. Scandurra, B. Pignataro, N. Gadegaard and L. Cronin, Angew. Chem., Int. Ed., 2009, 48, 6490–6493 CrossRef CAS; (g) J. Kang, B. Xu, Z. Peng, X. Zhu, Y. Wei and D. R. Powell, Angew. Chem., Int. Ed., 2005, 44, 6902–6905 CrossRef CAS; (h) C. P. Pradeep, M. F. Misdrahi, F. Y. Li, J. Zhang, L. Xu, D. L. Long, T. Liu and L. Cronin, Angew. Chem., Int. Ed., 2009, 48, 8309–8313 CrossRef CAS; (i) S. Noro, R. Tsunashima, Y. Kamiya, K. Uemura, H. Kita, L. Cronin, T. Akutagawa and T. Nakamura, Angew. Chem., Int. Ed., 2009, 48, 8703–8706 CrossRef CAS; (j) Y. Yan, H. Wang, B. Li, G. Hou, Z. Yin, L. Wu and V. W. W. Yam, Angew. Chem., Int. Ed., 2010, 49, 9233–9236 CrossRef CAS; (k) G. Maayan, R. Popovitz-Biro and R. Neumann, J. Am. Chem. Soc., 2006, 128, 4968–4969 CrossRef CAS; (l) P. P. Mishra, J. Pigga and T. Liu, J. Am. Chem. Soc., 2008, 130, 1548–1549 CrossRef CAS; (m) D. Li, J. Zhang, K. Landskron and T. Liu, J. Am. Chem. Soc., 2008, 130, 4226–4227 CrossRef CAS; (n) J. Zhang, Y. F. Song, L. Cronin and T. Liu, J. Am. Chem. Soc., 2008, 130, 14408–144409 CrossRef CAS.
- R. Neumann, Inorg. Chem., 2010, 49, 3594–3601 CrossRef CAS.
- J. T. Phule, W. A. Neiwert, K. I. Hardcastle, B. T. Do and C. L. Hill, J. Am. Chem. Soc., 2001, 123, 12101–12102 CrossRef CAS.
- F. X. Liu, C. Marchal-Roch, P. Bouchard, J. Marrot, J. P. Simonato, G. Herve and F. Secheresse, Inorg. Chem., 2004, 43, 2240–2242 CrossRef CAS.
- S. Kitagawa and S. Noro, in Comprehensive Coordination Chemistry II, ed. T. J. Meyer and J. A. McCleverty, Elsevier, Oxford, 2004, vol. 7, p. 231 Search PubMed.
- E. C. Constable, Aust. J. Chem., 2006, 59, 1–2 CrossRef CAS.
- A. Müller, E. Diemann, C. Kuhlmann, W. Eimer, C. Serain, T. Tak, A. Knöchel and P. K. Pranzas, Chem. Commun., 2001, 19, 1928–1929 Search PubMed.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. O. Bomben, Handbook of X-ray Photoelectron Spectroscopy, Perkin-Elmer Corporation, USA, 1992, pp. 120–121 Search PubMed.
- K. J. M. Bishop, C. E. Wilmer, S. Soh and B. A. Grzybowski, Small, 2009, 5, 1600–1630 CrossRef CAS.
- Y. S. Lee, Self-assembly and Nanotechnology: A Force Balance Approach, John Wiley & Sons, New Jersey, 2008, pp. 3–19 Search PubMed.
- (a) I. Cohen, T. G. Mason and D. A. Weitz, Phys. Rev. Lett., 2004, 93, 046001/1 Search PubMed; (b) N. B. Simeonova and W. K. Kegel, Phys. Rev. Lett., 2004, 93, 035701/1 Search PubMed; (c) P. K. Yuet, Langmuir, 2004, 20, 7960–7971 Search PubMed; (d) J. Zhu, M. Li, R. Rogers, W. Meyer and R. H. Ottewill, STS - 73 Space Shuttle Crew, W. B. Russel, P. M. Chaikin, Nature, 1997, 387, 883–885 Search PubMed.
- (a) M. Rycenga, J. M. McLellan and Y. Xia, Adv. Mater., 2008, 20, 2416–2420 CrossRef CAS; (b) M. M. Maye, D. Nykypanchuk, M. Cuisinier, D. van der Lelie and O. Gang, Nat. Mater., 2009, 8, 388–391 CrossRef CAS.
- Z. Nie, A. Petukhova and E. Kumacheva, Nat. Nanotechnol., 2010, 5, 15–25 CrossRef CAS.
- H. J. Arpe, Industrial Organic Chemistry, Wiley-VCH, Germany, 5th edn, 2010, pp. 397–416 Search PubMed.
- L. Lloyd, Handbook of Industrial Catalysts, Springer, UK, 2011, pp. 142–143 Search PubMed.
- S. Val, M. L. Granados, J. L. G. Fierro, J. Santamarıa-Gonzalez and A. Jimenez-Lopez, J. Catal., 1999, 188, 203–214 Search PubMed.
- (a) T. Forster, S. A. Schunk, A. Jentys and J. A. Lercher, J. Catal., 2011, 283, 25–33 Search PubMed; (b) T. Foster, S. A. Schunk, A. Jentys and J. A. Lercher Chem., Commun., 2011, 47, 3254–3256 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, Table S1, Table S2, and Fig. S1. See DOI: 10.1039/c2ra21858e |
| This journal is © The Royal Society of Chemistry 2012 |