Synthesis of carbohydrate-based naphthoquinones and their substituted phenylhydrazono derivatives as anticancer agents†
Vinicius R.
Campos
a,
Evelyne A. dos
Santos
b,
Vitor F.
Ferreira
a,
Raquel C.
Montenegro
c,
Maria C. B. V.
de Souza
a,
Letícia V.
Costa-Lotufo
b,
Manoel O.
de Moraes
b,
Anna K. P.
Regufe
a,
Alessandro K.
Jordão
a,
Angelo C.
Pinto
d,
Jackson A. L. C.
Resende
e and
Anna C.
Cunha
*a
aUniversidade Federal Fluminense, Departamento de Química Orgânica, Programa de Pós-Graduação em Química, Outeiro de São João Batista, 24020-141, Niterói, RJ, Brazil
bUniversidade Federal do Ceará, Departamento de Fisiologia e Farmacologia, Fortaleza, CE, Brazil
cUniversidade Federal do Pará, Instituto de Ciências Biológicas, Belém, PA, Brazil
dUniversidade Federal do Rio de Janeiro, Departamento de Química Orgânica, Instituto de Química-CT, Bloco A, Cidade Universitária, 21941-590, Rio de Janeiro, RJ, Brazil
eUniversidade Federal Fluminense, Departamento de Química Inorgânica, Laboratório Regional de Difração de Raios X (LDRX), 24020-141, Niterói, RJ, Brazil
First published on 25th September 2012
Abstract
A novel series of carbohydrate-based 1,2-naphthoquinones 13a–c and their substituted phenylhydrazono derivatives 14a–l were synthesized and evaluated for cytotoxicity against HL-60, MDA-MB435, HCT-116 and SF-295 cancer cell lines. The compounds 9a–c showed the best cytotoxicity profile (IC50 below 2 μM) against HL-60 and MDA-MB 435 human cells, while the hydrazone derivative 14i (IC50 HL-60 1.65 μM) was selective for leukemia when compared to the reference drug doxorubicin. None of the compounds exhibited lytic effects against mouse erythrocytes. Characterization of all compounds was confirmed by one- and two-dimensional NMR techniques (1H, 13C-APT, COSY-1H × 1H and HETCOR 1JCH) and by elemental analysis.
Introduction
The conjugation of carbohydrates with homo/heteroaromatic compounds has lead to the development of very effective anticancer drugs including the well-known clinical anticancer nucleoside analogues1 cladribine2 (1), clofarabine2–4 (2), fludarabine2,5 (3), gemcitabine2,3 (4) and decitabine2,6 (5) (Fig. 1). Nucleoside derivatives can exert their cytotoxicity using a range of mechanisms of action1,2 that result in inhibition of DNA synthesis and apoptotic cell death.Other examples of carbohydrate-based compounds (Fig. 1) that exhibit anticancer activity include bleomycin7,8 (BLM A5, 6), a drug clinically used in the treatment of squamous cell carcinomas and malignant lymphomas,9 and the naturally occurring alkaloid10 rebeccamycin (7), a DNA-damaging agent that inhibits the topoisomerase I enzyme. BLM A5 is a glycopeptide7 antibiotic that induces breakage of DNA molecules by oxidation of the deoxyribose moiety in the presence of oxygen and a metal ion, e.g., Fe or Co.11
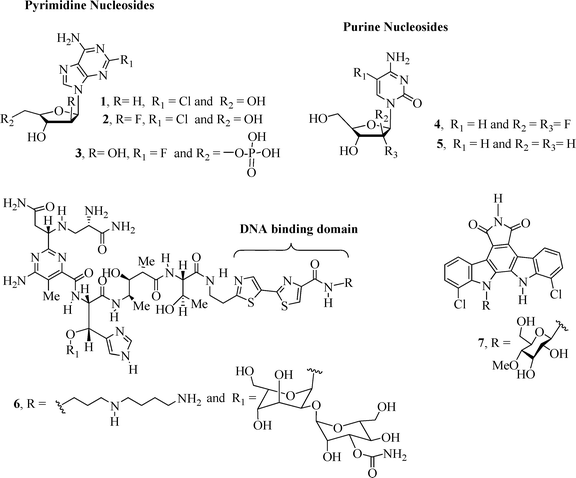 | ||
| Fig. 1 Examples of carbohydrate-based compounds 1–7 with anticancer activity. | ||
Many quinone derivatives are clinically important drugs used in the treatment of cancer.12,13 They also possess a variety of pharmacological effects14 including antimalarial,15 antifungal,16 and molluscicidal,17 as well as demonstrate significant activity against Trypanosoma cruzi18,19 and tuberculosis.20
Three major mechanisms have been proposed for the cytotoxic action of quinone derivatives in a variety of biological systems.12 These normally involve the generation of active oxygen species (e.g. superoxide radical anions, hydrogen peroxide and hydroxyl radicals) by redox cycling, intercalation in the DNA double helix or alkylation of biomolecules.12 The redox cycling of quinone derivatives may be initiated by either a one- or two-electron reduction. They can also be reduced by one-electron reductases to semiquinones (Q˙−), which can then undergo redox cycling in the presence of oxygen, generating superoxide anion radical (O2˙−). Superoxide can be converted to hydrogen peroxide (H2O2) via a superoxide dismutase (SOD)-catalysed reaction, followed by the formation of a hydroxyl radical (HO) by the iron-catalyzed reduction of peroxide via the Fenton reaction (Scheme 1). All of these oxygen intermediates or reactive oxygen species (ROS) may react directly with DNA or other biomolecules, such as lipids and proteins, leading to cell damage.12,21
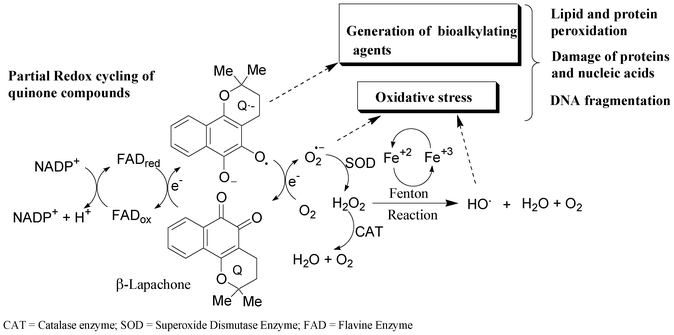 | ||
| Scheme 1 Formation of reactive oxygen species by redox cycling.12,21 | ||
Natural compounds containing quinone and carbohydrate moieties 8–12 possess significant antibiotic and antitumor activities (Fig. 2).22–26 In particular, anthracycline antibiotics 10–12 are known to be some of the most effective drugs against a wide variety of solid tumors in human patients. The mechanisms26–29 of activity of these clinically active drugs interferes with cell growth and proliferation, resulting in an inhibition of DNA synthesis and apoptotic cell death. The sugar substructure attached to the quinone ring is an important structural requirement for the bioactivity of these anthracycline antibiotics;30–33 the moiety participates in molecular recognition of the main cellular target DNA and topoisomerase II, forming a DNA–drug–topoisomerase II ternary complex in which the enzyme is covalently linked to the broken DNA strand. This event is critical for inducing apoptosis and cell death.30–33
Quinones such as shikonin, an active component isolated from the traditional medicinal herb Lithospermium erythrorhizon, exhibits anticancer activity by inhibiting telomerase and DNA topoisomerase I/II, and cancer cell growth. Recently, shikonin derivatives attached to different carbohydrate groups were reported to exhibit superior cytotoxicity and solubility when compared with unglycosylated drug.34
Schiff bases containing azomethine-NHN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH protons have been used in the preparation of many potential drugs and exhibit a broad spectrum of biological activities such as antiviral,35 antibacterial,36 anti-inflammatory,37 antiplatelet38 and anticancer39 activities. Notably, the introduction of a hydrazone group onto the naphthoquinone40,41 and aryl42 nucleus has led to promising anticancer agents.
CH protons have been used in the preparation of many potential drugs and exhibit a broad spectrum of biological activities such as antiviral,35 antibacterial,36 anti-inflammatory,37 antiplatelet38 and anticancer39 activities. Notably, the introduction of a hydrazone group onto the naphthoquinone40,41 and aryl42 nucleus has led to promising anticancer agents.
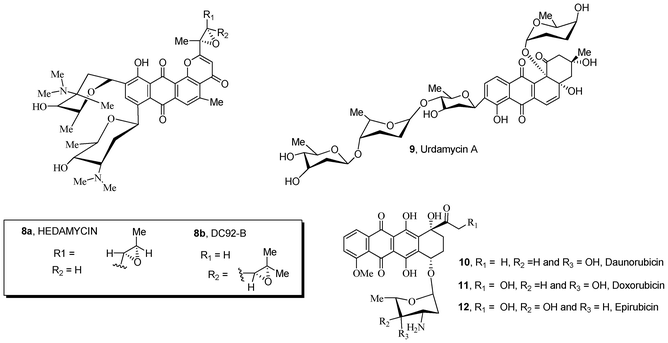 | ||
| Fig. 2 Natural sugar-containing quinones 8–12 with anticancer activity. | ||
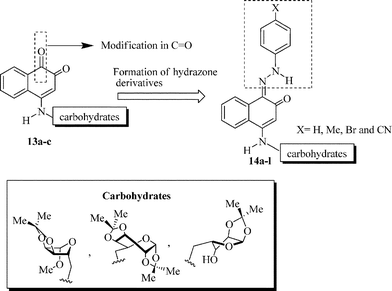
In this context, the synthesis of carbohydrate-containing quinone derivatives is currently an important field of pharmacy and chemistry. Herein, we report the synthesis and in vitro antitumor activity of a new family of sugar-based 1,2-naphthoquinones 13a–c (Scheme 2). We also investigate the contribution of different hydrazone-substituted phenyl rings attached to the quinone moiety, probing the biological activity of a new series of hydrazone-containing naphthoquinone derivatives 14a–l.
 | ||
| Scheme 2 Strategy for the synthesis of compounds 13a–c and 14a–l. | ||
The cytotoxicity of the new compounds 13a–c and 14a–l was evaluated against human cancer cell lines including leukemia (HL-60), melanoma (MDA-MB 435), colon (HCT-116) and glioblastoma (SF-295).
Results and discussion
Methyl 5-amino-5-deoxy-1,2-O-isopropylidene-β-D-ribofuranoside 15a, 6-amino-1,2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3,4-di-O-isopropylidene-α-D-galactopyranose 15b and 5-amino-5-deoxy-1,2-O-isopropylidene-α-D-xylofuranose 15c were prepared from the corresponding commercially available reagents D-ribose, D-xylose and D-galactose using known methods of carbohydrate derivatization.14 The reaction of sodium 1,2-naphthoquinone-4-sulfonate (16) with aminocarbohydrates 15a–c led to the formation of the 4-amino-1,2-naphthoquinone derivatives, as illustrated in Scheme 3. Table 1 shows the melting points of these compounds and the reaction yields.
:3,4-di-O-isopropylidene-α-D-galactopyranose 15b and 5-amino-5-deoxy-1,2-O-isopropylidene-α-D-xylofuranose 15c were prepared from the corresponding commercially available reagents D-ribose, D-xylose and D-galactose using known methods of carbohydrate derivatization.14 The reaction of sodium 1,2-naphthoquinone-4-sulfonate (16) with aminocarbohydrates 15a–c led to the formation of the 4-amino-1,2-naphthoquinone derivatives, as illustrated in Scheme 3. Table 1 shows the melting points of these compounds and the reaction yields.
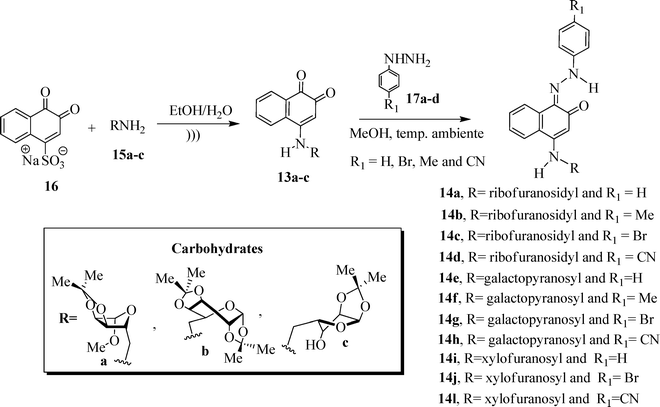 | ||
| Scheme 3 Synthetic pathways used to prepare 13a–c and 14a–l. | ||
| Compound | R | R1 | Mp/°C | Yield (%) |
|---|---|---|---|---|
| 13a | Ribofuranosidyl | — | 195–198 | 65 |
| 13b | Galactopyranosyl | — | 215–218 | 50 |
| 13c | Xylofuranosyl | — | 208–210 | 55 |
| 14a | Ribofuranosidyl | H | 170–172 | 55 |
| 14b | Ribofuranosidyl | Me | 155–157 | 60 |
| 14c | Ribofuranosidyl | Br | 201–203 | 60 |
| 14d | Ribofuranosidyl | CN | 247–249 | 55 |
| 14e | Galactopyranosyl | H | 95–97 | 55 |
| 14f | Galactopyranosyl | Me | 105–107 | 50 |
| 14g | Galactopyranosyl | Br | 110–112 | 55 |
| 14h | Galactopyranosyl | CN | 223–225 | 50 |
| 14i | Xylofuranosyl | H | 205–207 | 55 |
| 14j | Xylofuranosyl | Br | 220–221 | 50 |
| 14l | Xylofuranosyl | CN | 230–232 | 40 |
The structures of 4-aminonaphthoquinones 13a–c were confirmed by one- and two-dimensional NMR techniques (1H, 13C-APT, COSY-1H × 1H and HETCOR 1JCH) as well as elemental analysis. The envelope (E) and twist (T) conformations for compounds 13a and 13c, respectively, were established based on the proton coupling constants (J) of the hydrogen signals of the furan rings and by X-ray crystallographic data of the molecular structures of compounds 14c and 14j (Fig. 3 and 4).
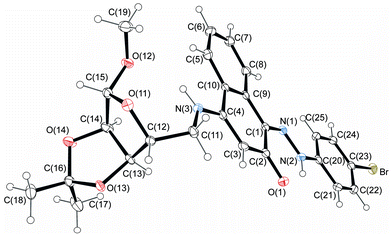 | ||
| Fig. 3 Molecular structure of compound 14c. Displacement ellipsoids for non-hydrogen atoms are drawn at the 30% probability level. | ||
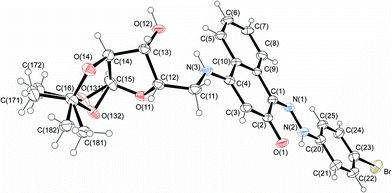 | ||
| Fig. 4 Molecular structure of compound 14j, with disorder in the acetonide rings. Displacement ellipsoids for non-hydrogen atoms are drawn at the 30% probability level. | ||
The twist-boat conformation of the D-galactose ring in 13b was confirmed by the 1H–1H vicinal coupling constant values JH-1′,H-2′, JH-2′,H-3′, JH-3′,H-4′ and JH-4′,H-5′ (5.0, 2.1, 7.8 and 1.5 Hz, respectively) and by hydrogen chemical shift comparison with literature data for the analogous carbohydrates.43
The reaction of 1,2-naphthoquinone derivatives 13a–c with substituted phenylhydrazine hydrochlorides 17a–d led to the corresponding hydrazone derivatives, as illustrated in Scheme 1. Although the possibility of the nucleophilic attack of hydrazine derivatives on the carbonyl carbon atom C-1 or C-2 of 13a–c, only hydrazone compounds 14a–l were isolated, resulting from the nucleophilic addition to the more electrophilic carbonyl group (C-1).
In this type of reaction, E- and Z-hydrazones are in equilibrium through the intermediate hemiaminals 18a–l (Scheme 4).
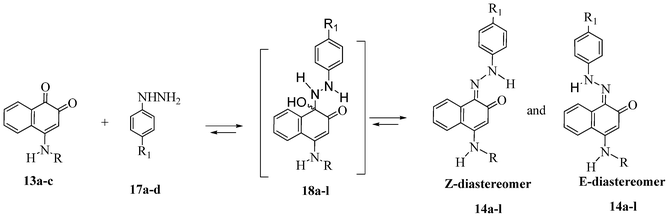 | ||
| Scheme 4 Equilibrium of E- and Z-hydrazones through intermediate hemiaminals 18a–l. | ||
The 1H NMR spectra of 14a–l indicate that only one of two possible isomers was formed. The (Z)-stereochemistry for the CN imine double bond in the series of the hydrazono derivatives 14a–l was assigned on the basis of X-ray crystallographic analysis of the molecular structures of 14c and 14j (Fig. 3 and 4). The (Z)-conformation of 14c and 14j was established by the intramolecular N(2)–H(2A)⋯O(1) six-membered hydrogen-bonded ring and the π-electron delocalization in the adjacent aromatic systems. Furthermore, the bond length C(1)–N(1) is the shortest distance between the C and N atoms in 14c and 14j, indicating a double bond. In both structures, the hydrazone chain and quinone ring form a planar system, with bond distances typical of delocalized aromatic compounds.
The carbohydrate chain in compound 14c adopts an envelope conformation in which the O(11) atom is located at 0.300(3) Å, out of the plane of the other four atoms C(12), C(13), C(14) and C(15), with Cremer–Pople puckering parameters q2 and ϕ2 of 0.22(1) Å and 27(2)°, respectively. The furan sugar ring of 14j has a twist conformation, ϕ2 = 20(2)°; q2 = 0.39(1) Å, according to Cremer–Pople puckering parameters, in which two adjacent C(12) and C(13) carbon atoms lie above [0.313(16) Å] and below [−0.340(17) Å] the plane defined by the remaining three atoms, respectively.
Structurally important interactions are summarized in Table 2. For example, in both crystals, N(2)–H(2A)⋯O(1) and N(3)–H(3A)⋯O(12) intramolecular hydrogen bonds are observed. Analysis of the crystal packing for compounds 14c and 14j reveals a net of weak C–H⋯O hydrogen bonds and π⋯π stacking interactions. Additionally, intermolecular hydroxyl group hydrogen bonds in 14j form an chain along the b-axis.
| D–H⋯A | d(D–H) | d(H⋯A) | d(D⋯A) | θ(D–H⋯A) | Symmetry code | |
|---|---|---|---|---|---|---|
| 14c | N(2)–H(2A)⋯O(1) | 0.88 | 1.86 | 2.552(2) | 135 | — |
| N(3)–H(3A)⋯O(12) | 0.88 | 2.15 | 2.952(3) | 151 | — | |
| C(7)–H(7)⋯O(14)i | 0.95 | 2.40 | 3.251(3) | 149 | (i) 1 − x, ½ − y, 1½ + z | |
| C(11)–H(11B)⋯O(1)ii | 0.98 | 2.47 | 3.212(3) | 132 | (ii) ½ + x, ½ − y, 2 − z | |
| C(17)–H(17B)⋯O(11)iii | 0.98 | 2.43 | 3.399(3) | 169 | (iii) −1 + x, y, z | |
| 14j | N(2)–H(2A)⋯O(1) | 0.88 | 1.83 | 2.536(7) | 136 | — |
| N(3)–H(3A)⋯O(12) | 0.88 | 2.03 | 2.713(7) | 133 | — | |
| O(12)–H(12O)⋯O(1)iv | 0.84 | 1.84 | 2.645(6) | 160 | (iv) −x, −½ + y, ½ − z | |
| C(7)–H(7)⋯O(12)iv | 0.98 | 2.54 | 3.276(8) | 136 | (v) 1 − x, −½ + y, ½ − z | |
| C(15)–H(15)⋯O(14)iii | 0.98 | 2.34 | 3.155(7) | 141 | (iii) 1 + x, y, z | |
Biological assay
The in vitro anticancer activities of the new carbohydrate-based naphthoquinones 13a–c and the phenylhydrazono derivatives 14a–l were assessed against four human cancer cell lines in comparison to doxorubicin (positive control) using the MTT assay. The concentrations that induce 50% inhibition of cell growth (IC50) are reported in Table 3 in μg mL−1 (μM).| Cancer cell line | ||||
|---|---|---|---|---|
| Compound | HL-60 | MDA-MB-435 | HCT-116 | SF-295 |
| a 95% confidence intervals were obtained by nonlinear regression for all cell lines from three independent experiments (IC50/μg mL−1). Doxorubicin (DOX) was used as positive control. Nd– not determined. | ||||
| 13a | 1.27 (3.53) | 0.87 (2.42) | 4.00 (11.13) | 2.65 (7.37) |
| 0.73–2.19a | 0.69–1.08a | 3.66–4.37a | 2.18–3.22a | |
| 13b | 1.83 (4.40) | 1.13 (2.72) | >5 (12.03) | 2.17 (5.22) |
| 1.01–3.30a | 0.86–1.48a | 1.88–2.49a | ||
| 13c | 1.29 (3.73) | 1.97 (5.70) | 3.22 (9.32) | 2.34 (6.77) |
| 0.64–2.59a | 1.52–2.54a | 2.66–3.89a | 1.89–2.90a | |
| 14a | >5 (11.12) | >5 (11.12) | >5 (11.12) | >5 (11.12) |
| 14b | >5 (10.79) | >5 (10.79) | >5 (10.79) | >5 (10.79) |
| 14c | >5 (9.46) | >5 (9.46) | >5 (9.46) | >5 (9.46) |
| 14d | >5 (10.54) | >5 (10.54) | >5 (10.54) | >5 (10.54) |
| 14e | >5 (9.89) | >5 (9.89) | >5 (9.89) | >5 (9.89) |
| 14f | >5 (9.62) | >5 (9.62) | >5 (9.62) | >5 (9.62) |
| 14g | >5 (8.55) | >5 (8.55) | >5 (8.55) | >5 (8.55) |
| 14h | >5 (9.42) | >5 (9.42) | >5 (9.42) | >5 (9.42) |
| 14i | 1.65 (3.79) | >5 | >5 (11.48) | 4.48 (10.29) |
| 1.03–2.65a | 3.78–5.30a | |||
| 14j | >5 (9.72) | >5 (9.72) | >5 (9.72) | >5 (9.72) |
| 14l | >5 (9.42) | >5 (9.42) | >5 (9.42) | >5 (9.42) |
| Doxorubicin | 0.03 (0.5) | 0.88 (1.52) | Nd | 0.41 (0.71) |
| 0.02–0.04a | 0.62–1.21a | 0.29–0.44a | ||
The results indicate that naphthoquinone derivatives 13a–c, which were attached to various carbohydrate groups, exhibited a considerable cytotoxic effect against all cancer cell lines and without any lytic effect against erythrocytes. These results are in accordance with National Cancer Institute (NCI) protocols, where compounds exhibiting IC50 values <4 μg ml−1 and below 10 μM are considered active.44
The data collected from three independent experiments, in triplicate, reveal that 13a, 4-aminonaphthalene-1,2-dione, is the most active derivative, with IC50 value of 0.87 μg ml−1 against the melanoma cell line (MDA-MB-435). The enhanced anticancer activity of 13a can be related to the chemical structure (e.g., conformation and intermolecular interactions) of the pyranose ring. We also speculate that cellular permeability of the 1,2-naphthoquinonic system can be improved with the construction of the new compounds 13a–c possessing a carbohydrate chain at C-4 position of the quinone moiety.
Compounds 13a,b were slightly less active than the clinically anticancer agent doxorubicin against all cancer cell lines tested. Although doxorubicin be considered a mainstay of cancer chemotherapy,45 it has several clinical limitations such as cardiotoxic effects and a high incidence of multi-drug resistance.
Among the hydrazones 14a–l, only the unsubstituted hydrazone derivative 14i showed significant activity against leukemia cells lines. A dramatic loss of cytotoxicity for these compounds compared to the cytotoxic activity of their precursors 13 can be related to the absence of the redox center group CO, which was derivatized as a hydrazone moiety (–CN–NH–), leading to lower toxicity.
Non-substituted hydrazone compound 14i showed selective cytotoxicity against HL-60 cell lines, with IC50 value of 1.65 μg ml−1, whereas IC50 values over 5 μg ml−1 were observed against all other cell lines. These results suggest that the compound 14i can act as a prodrug of 13c improving its antitumor selectivity. Studies have been reported the use of hydrazone prodrugs as an important approach for enhancing the antitumor selectivity and decreasing severe side effect of antitumor agents.46 The hydrazone linker is relatively stable at neutral pH, but labile under acidic conditions.47 According to literature, this class of the compound can be metabolized in vitro and in vivo to biologically active carbonyl compounds.46
The mechanical stability of red blood cells is a good parameter for in vitro screening of unspecific cytotoxicity because erythrocyte membranes can tolerate significant structural change.48 Assays of compounds 13a–c and 14a–l showed no hemolytic activity (EC > 250 mg mL−1).We therefore suggest that the toxicity mechanism involved is not related to nonspecific membrane damage (Table 3).
Conclusion
In summary, a new series of naphthoquinones 13a–c and their corresponding phenylhydrazones 14a–l have been synthesized and evaluated for anticancer activity against human cancer cell lines including leukemia (HL-60), melanoma (MDA-MB-435), colon (HCT-116) and central nervous system (SF-295). Compounds 13a–c exhibited considerable cytotoxic activity. The compound 4-((methyl-5′-deoxy-2′,3′-O-isopropylidene-β-D-methylfuranosid-5′-yl)methylamino)naphthalene-1,2-dione (13a) was the most active of this series, with IC50 values below 2 μg ml−1 against leukemia and melanoma cell lines.Except for compound (Z)-4-((5′-deoxy-1,2-O-isopropylidene-α-D-xylofuranosid-5′-yl)methylamino)-1-(2-phenyl)hydrazono)naphthalene-2(1H)-one (14i), the introduction of a hydrazone pharmaphoric group at the C-1 position of naphthoquinone ring led to a dramatic drop in antitumor activity against all tested cancer cell lines. Compound 14i displayed an IC50 of 1.65 μg ml−1 against leukemia cells (HL-60), whereas doxorubicin was not selective against any cancer cell line. These pharmacology results suggest that the compound 14i can act as an anticancer prodrug of 13c improving its antitumor selectivity. No compounds exhibited lytic effects on mouse erythrocytes.
Due to the in vitro anticancer activity of these novel naphthoquinone derivatives 13a–c and 14i, we believe that these compounds are attractive for further structural modifications contributing to the design of new antiproliferative agents.
Experimental
Chemical reagents and solvents used in this study were purchased from Merck AG (Darmstadt, Germany) and VETEC LTDA. The synthesis of new compounds 13a–c and 14a–l was performed in a 40-kHz ultrasonic bath (USC 1400, ULTRASONIC CLEANER, UNIQUE, Indaiatuba, São Paulo, Brazil). Melting points were determined with a Fisher-Johns instrument and are uncorrected. Infrared (IR) spectra were recorded on a Perkin-Elmer FT-IR 1600 spectrophotometer using KBr pellets. NMR spectra, unless otherwise stated, were obtained in deuterated DMSO and CDCl3 using Varian Unity Plus 300 and 500 MHz spectrometers. Chemical shifts (δ) are expressed in ppm and coupling constants (J) in Hertz. Optical rotation was recorded with a Perkin-Elmer 243B Polarimeter (sodium lamp at 589 nm). Microanalyses were performed on a Perkin-Elmer 2400 instrument, and all reported values were within ±0.4% of the calculated compositions.The progress of all reactions was monitored by TLC performed on 2.0 cm × 6.0 cm aluminium sheets pre-coated with silica gel 60 (HF-254, E. Merck) to a thickness of 0.25 mm. The developed chromatograms were viewed under ultraviolet light at 254 nm. Merck silica gel (60–200 mesh) was used for column chromatography.
4.1. General procedure for the preparation of the 4-aminonaphthalene-1,2-dione derivatives 13a–c
A solution of 1,2-naphthoquinone-4-sulfonic acid sodium salt (16, 1.0 mmol) and aminocarbohydrate 15a–c (1.0 mmol) in 10 mL EtOH–H2O (1:1) was sonicated for 1 h at room temperature. The solvent was removed under reduced pressure. The residue was purified by flash column chromatography (gradient elution, 10–30% EtOAc in hexane) to afford the desired compounds 13a–c.
O).
1H NMR (300.00 MHz, DMSO) δ 1.25 (s, 3H, CH3), 1.37 (s, 3H, CH3), 3.28 (s, 3H, OCH3), 3.46–3.52 (m, 2H, H-5′ and H-5′′), 4.43 (dd, 1H, J = 6.9 and 7.8 Hz, H-4′), 4.66 (d, 1H, J = 6.0 Hz, H-2′), 4.83 (d, 1H, J = 6.0 Hz, H-3′), 4.98 (s, 1H, H-1′), 5.67 (s, 1H, H-3), 7.70 (td, J = 1.5 and 7.8 Hz, H-6), 7.84 (td, J = 1.5 and 7.8 Hz, H-7), 8.0 (dd, J = 1.5 and 7.8 Hz, H-8), 8.15 (dd, J = 1.5 and 7.8 Hz, H-5), 8.40 (br s, 1H, N–H).
13C NMR (75.0 MHz, DMSO) δ 24.6 (CH3), 26.1 (CH3), 46.0 (C-5′), 54.4 (OCH3), 81.7 (C-2′), 82.9 (C-4′), 84.4 (C-3′), 98.2 (C-3), 108.7 (C-1′), 111.5 (-OCO-), 123.3 (C-8), 127.9 (C-5), 130.0 (C-4a), 131.3 (C-6), 131.0 (C-8a), 134.2 (C-7), 154.7 (C-4), 174.5 (C-2), 181.5 (C-1).
Anal. Calc. for C19H21NO6: C, 63.50; H, 5.89; N, 3.90. Found: C, 62.92; H, 6.04; N, 3.91%.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :3′,4′-di-O-isopropylidene-D-galactopiranos-6′-yl)methylamino)naphthalene-1,2-dione (13b).
The reaction between naphthoquinone 16 (260 mg, 1 mmol) and aminocarbohydrate 15b (259 mg, 1.0 mmol) yielded 13b (207 mg, 0.5 mmol, 50%) as an orange solid: mp 215–218 °C; [α]D20 −15 (c 0.002, CH2Cl2); IR (KBr) ν/cm−1: 3305 (N–H), 1694 and 1591 (CO).
:3′,4′-di-O-isopropylidene-D-galactopiranos-6′-yl)methylamino)naphthalene-1,2-dione (13b).
The reaction between naphthoquinone 16 (260 mg, 1 mmol) and aminocarbohydrate 15b (259 mg, 1.0 mmol) yielded 13b (207 mg, 0.5 mmol, 50%) as an orange solid: mp 215–218 °C; [α]D20 −15 (c 0.002, CH2Cl2); IR (KBr) ν/cm−1: 3305 (N–H), 1694 and 1591 (CO).
1H NMR (300.00 MHz, DMSO) δ 1.21 (s, 3H, CH3), 1.28 (s, 3H, CH3), 1.32 (s, 3H, CH3), 1.41 (s, 3H, CH3), 3.76–3.86 (m, 2H, H-6′ and H-6′′), 4.10–4.13 (m, 1H, H-5′), 4.34 (dd, 1H, J = 2.1 and 5.0 Hz, H-2′), 4.40 (dd, 1H, J = 1.5 and 7.8 Hz, H-4′), 4.63 (dd, 1H, J = 2.1 and 7.8 Hz, H-3′), 5.45 (d, 1H, J = 5.0 Hz, H-1′), 5.77 (s, 1H, H-3), 7.68 (td, 1H, J = 1.5 and 7.5 Hz, H-6), 7.82 (td, 1H, J = 1.5 and 7.5 Hz, H-7), 7.98 (dd, 1H, J = 1.5 and 7.5 Hz, H-5), 8.11 (dd, 1H, J = 1.5 and 7.5 Hz, H-8), 8.56 (m, 1H, N–H).
13C NMR (75.0 MHz, DMSO) δ 24.2 (CH3), 24.8 (CH3), 25.6 (CH3), 26.0 (CH3), 42.9 (C-6′), 64.3 (C-5′), 70.1 (C-3′), 70.5 (C-4′), 79.8 (C-2′), 95.6 (C-1′), 98.5 (C-3), 108.0 (C-7′), 108.5 (C-8′), 123.1 (C-8), 127.8 (C-5), 130.8 (C-4a), 131.1 (C-8a), 131.3 (C-6), 134.3 (C-7), 154.9 (C-4), 174.7 (C-2), 181.8 (C-1).
Anal. Calc. for C22H25NO7: C, 63.60; H, 6.07; N, 3.37. Found: C, 63.14; H, 6.19; N, 3.33%.
O).
1H NMR (300.00 MHz, DMSO) δ 1.25 (s, 3H, CH3), 1.36 (s, 3H, CH3), 3.50–3.58 (m, 1H, H-5′′), 3.64–3.74 (m, 1H, H-5′), 4.12–4.16 (m, 1H, H-3′), 4.34–4.40 (m, 1H, H-4′), 4.46 (d, 1H, J = 3.6 Hz, H-2′), 5.54 (1H; d; J = 4.8 Hz, OH), 5.77 (s, 1H, H-3), 5.87 (d, 1H, J = 3.6 Hz, H-1′), 7.67 (td, J = 1.2 and 7.5 Hz, H-6), 7.80 (td, J = 1.2 and 7.5 Hz, H-7), 7.97 (dd, J = 1.2 and 7.5 Hz, H-5), 8.10 (dd, J = 1.2 and 7.5 Hz, H-8), 8.40 (dd, 1H, J = 8.4 Hz, N–H).
13C NMR (75.0 MHz, DMSO) δ 25.9 (CH3), 26.6 (CH3), 42.2 (C-5′), 77.8 (C-4′), 76.6 (C-3′), 85.0 (C-2′), 98.2 (C-3), 104.2 (C-1′), 110.6 (C-6′), 123.3 (C-8), 127.8 (C-5), 130.7 (C-4a), 131.1 (C-8a), 131.3 (C-6), 134.2 (C-7), 155.0 (C-4), 174.9 (C-2), 181.5 (C-1).
Anal. Calc. for C18H19NO6: C, 62.60; H, 5.55; N, 4.06. Found: C, 61.94; H, 6.21; N, 4.53%.
4.2. General procedure for the preparation of the phenylhydrazone derivatives 14a–l
A mixture of 4-aminocarbohydrate-1,2-naphthoquinone (1.0 mmol, 13a–c) and phenylhydrazine hydrochloride (1.0 mmol, 17a–d) in MeOH (10 mL) was stirred for 24 h at room temperature. The reaction mixture was concentrated under reduced pressure and the resulting residue was purified by column chromatography using silica gel and ethyl acetate–hexane (3:7) as the eluent to give the pure phenylhydrazone derivatives 14a–l.
O), 3431 (N–H) and 1600 (CN).
1H NMR (300.00 MHz, CDCl3) δ 1.33 (s, 3H, CH3), 1.52 (s, 3H, CH3), 3.44 (s, 3H, OCH3), 3.46–3.49 (m, 2H, H-5′ and H-5′′), 4.68 (td, 1H, J = 0.6 and 4.5 Hz, H-4′), 4.72 (d, 1H, J = 6.0 Hz, H-2′), 4.77 (dd, 1H, J = 0.6 and 6.0 Hz, H-3′), 5.12 (s, 1H, H-1′), 5.82 (s, 1H, H-3), 6.32 (br s, 1H, N–H), 7.10 (tt, 1H, J = 1.2 and 7.8 Hz, H-4′′), 7.36–7.41 (m, 3H, H-3′′, H-5′′ and H-6), 7.51–7.56 (m, 3H, H-2′′, H-6′′ and H-7), 7.60 (dd, 1H, J = 1.2 and 7.8 Hz, H-5), 8.51 (dd, 1H, J = 1.2 and 7.8 Hz, H-8).
13C NMR (75.0 MHz, CDCl3) δ 24.8 (CH3), 26.4 (CH3), 46.1 (C-5′), 55.6 (OCH3), 82.1 (C-2′), 85.6 (C-3′), 85.9 (C-4′), 100.0 (C-3), 110.1 (C-1′), 112.8 (C-6′), 115.8 (C-2′′ and C-6′′), 120.2 (C-5), 122.9 (C-4a), 123.0 (C-8), 124.0 (C-4′′), 126.1 (C-6), 128.1 (C-1), 129.4 (C-3′′ and C-5′′), 129.5 (C-7), 134.7 (C-8a), 143.1 (C-1′′), 153.6 (C-4), 179.4 (C-2).
Anal. Calc. for C25H27N3O5: C, 66.80; H, 6.05; N, 9.35. Found: C, 66.81; H, 6.12; N, 9.03%.
O), 3430 (N–H) and 1602 (CN).
1H NMR (500.00 MHz, CDCl3) δ 1.33 (s, 3H, CH3), 1.52 (s, 3H, CH3), 2.36 (s, 3H, Ar–CH3), 3.45 (s, 3H, OCH3), 3.49–3.52 (m, 2H, H-5′ and H-5′′), 4.66–4.69 (m, 1H, H-4′), 4.72 (d, 1H, J = 6.0 Hz, H-2′), 4.77 (d, 1H, J = 6.0 Hz, H-3′), 5.13 (s, 1H, H-1′), 5.96 (s, 1H, H-3), 6.40 (br s, 1H, N–H), 7.20 (d, 2H, J = 8.0 Hz, H-3′′ and H-5′′), 7.40 (td, 1H, J = 1.5 and 8.0 Hz, H-6), 7.44 (d, 2H, J = 8.0 Hz, H-2′′ and H-6′′), 7.55 (td, 1H, J = 1.5 and 8.0 Hz, H-7), 7.61 (dd, 1H, J = 1,5 and 8.0 Hz, H-5), 8.51 (dd, 1H, J = 1.5 and 8.0 Hz, H-8), 16.0 (s, 1H, N–N–H).
13C NMR (125.0 MHz, CDCl3) δ 20.9 (Ar–CH3), 24.8 (CH3), 26.4 (CH3), 46.1 (C-5′), 55.6 (OCH3), 82.2 (C-2′), 85.6 (C-3′), 85.7 (C-4′), 100.0 (C-3), 110.1 (C-1′), 112.8 (C-6′), 115.8 (C-2′′ and C-6′′), 120.2 (C-5), 122.7 (C-4a), 123.0 (C-8), 125.8 (C-6), 128.1 (C-1), 129.4 (C-7), 130.0 (C-3′′ and C-5′′), 134.0 (C-4′′), 134.7 (C-8a), 140.7 (C-1′′), 153.4 (C-4), 178.7 (C-2).
Anal. Calc. for C26H29N3O5: C, 67.37; H, 6.31; N, 9.07. Found: C, 67.16; H, 6.44; N, 8.88%.
O), 3340 (N–H) and 1591 (CN).
1H NMR (300.00 MHz, CDCl3) δ 1.33 (s, 3H, CH3), 1.53 (s, 3H, CH3), 3.45 (s, 3H, OCH3), 3.47–3.50 (m, 2H, H-5′ and H-5′′), 4.68 (td, 1H, J = 0.6 and 4.0 Hz, H-4′), 4.72 (d, 1H, J = 6.0 Hz, H-2′), 4.76 (dd, 1H, J = 0.6 and 6.0 Hz, H-3′), 5.12 (s, 1H, H-1′), 5.81 (s, 1H, H-3), 6.37 (br s, 1H, N–H), 7.37–7.43 (m, 1H, H-6), 7.39 (d, 2H, J = 8.4 Hz, H-2′′ and H-6′′), 7.49 (d, 2H, J = 8.4 Hz, H-3′′ and H-5′′), 7.58 (td, 1H, J = 1.2 and 8.1 Hz, H-7), 7.60 (dd, 1H, J = 1.2 and 8.1 Hz, H-5), 8.46 (dd, 1H, J = 1.2 and 8.1 Hz, H-8).
13C NMR (75.0 MHz, CDCl3) δ 24.8 (CH3), 26.4 (CH3), 46.2 (C-5′), 55.6 (OCH3), 82.1 (C-2′), 85.4 (C-4′), 85.5 (C-3′), 99.8 (C-3), 110.1 (C-1′), 112.9 (C-6′), 116.5 (C-4′′), 117.3 (C-2′′ and C-6′′), 120.4 (C-5), 123.1 (C-4a), 123.2 (C-8), 126.6 (C-6), 128.7 (C-1), 129.9 (C-7), 132.4 (C-3′′ and C-5′′), 134.0 (C-8a), 142.0 (C-1′′), 154.5 (C-4), 178,3 (C-2).
Anal. Calc. for C25H26BrN3O5: C, 56.83; H, 4.96; N, 7.95. Found: C, 56.81; H, 5.41; N, 7.56%.
O), 3380 (N–H), 1594 (CN) and 2221 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N).
N).
1H NMR (300.00 MHz, CDCl3) δ 1.33 (s, 3H, CH3), 1.52 (s, 3H, CH3), 3.46 (s, 3H, OCH3), 3.52–3.55 (m, 2H, H-5′ and H-5′′), 4.67–4.69 (m, 1H, H-4′), 4.72 (d, 1H, J = 6.0 Hz, H-2′), 4.75 (d, 1H, J = 6.0 Hz, H-3′), 5.13 (s, 1H, H-1′), 5.98 (s, 1H, H-3), 6.71 (br s, 1H, N–H), 7.46 (td, 1H, J = 1.0 and 8.0 Hz, H-6), 7.53 (d, 2H, J = 9.0 Hz, H-2′′ and H-6′′), 7.59 (td, 1H, J = 1.2 and 8.0 Hz, H-7), 7.61 (dd, 1H, J = 1.0 and 8.0 Hz, H-5), 7.65 (d, 2H, J = 9.0 Hz, H-3′′ and H-5′′), 8.46 (dd, 1H, J = 1.0 and 8.0 Hz, H-8), 15.72 (N–N–H).
13C NMR (75.0 MHz, CDCl3) δ 24.8 (CH3), 26.4 (CH3), 46.1 (C-5′), 54.8 (OCH3), 82.1 (C-2′), 85.5 (C-4′), 85.6 (C-3′), 99.7 (C-3), 105.7 (CN), 110.1 (C-1′), 112.9 (C-6′), 115.5 (C-2′′ and C-6′′), 119.4 (C-4′′), 120.4 (C-5), 123.6 (C-8), 123.8 (C-4a), 127.2 (C-6), 130.0 (C-7), 130.8 (C-1), 133.6 (C-3′′ and C-5′′), 134.1 (C-8a), 146.6 (C-1′′), 154.6 (C-4), 179.9 (C-2).
Anal. Calc. for C26H26N4O5: C, 65.81; H, 5.52; N, 11.81. Found: C, 65.98; H, 6.27; N, 11.47%.
O), 3423 (N–H) and 1593 (CN).
1H NMR (500.00 MHz, CDCl3) δ 1.35 (s, 3H, CH3), 1.39 (s, 3H, CH3), 1.50 (s, 3H, CH3), 1.53 (s, 3H, CH3), 3.50–3.62 (m, 2H, H-6′ and H-6′′), 4.15-4.17 (m, 1H, H-5′), 4.33 (dd, 1H, J = 2.5 and 8.0 Hz, H-4′), 4.37 (dd, 1H, J = 2.5 and 4.5 Hz, H-2′), 4.67 (dd, 1H, J = 2.5 and 8.0 Hz, H-3′), 5.60 (d, 1H, J = 4.5 Hz, H-1′), 5.87 (s, 1H, H-3), 5.98 (m, 1H, N–H), 7.10 (tt, 1H, J = 1.2 and 7.5 Hz, H-4′′), 7.37–7.40 (m, 3H, H-6, H-3′′ and H-5′′), 7.48–7.55 (m, 4H, H-5, H-7, H-2′′ and H-6′′), 8.50 (dd, 1H, J = 1.5 and 8.0 Hz, H-8).
13C NMR (125.0 MHz, CDCl3) δ 24.2 (CH3), 24.9 (CH3), 26.0 (CH3), 26.1 (CH3), 44.1 (C-6′), 65.1 (C-5′), 70.6 (C-2′), 70.9 (C-3′), 72.0 (C-4′), 96.4 (C-1′), 99.0 (C-3), 108.9 (C-7′), 109.7 (C-8′), 115.8 (C-2′′ and C-6′′), 120.2 (C-5), 122.6 (C-4a), 123.1 (C-8), 124.1 (C-4′′), 126.1 (C-6), 127.8 (C-1), 129.4 (C-3′′ and C-5′′), 129.7 (C-7), 134.7 (C-8a), 143.0 (C-1′′), 153.7 (C-4), 178.1 (C-2).
Anal. Calc. for C28H31N3O6: C, 66.52; H, 6.18; N, 8.31. Found: C, 66.58; H, 6.35; N, 7.71%.
O), 3420 (N–H) and 1591 (CN).
1H NMR (300.00 MHz, CDCl3) δ 1.34 (s, 3H, CH3), 1.40 (s, 3H, CH3), 1.50 (s, 3H, CH3), 1.53 (s, 3H, CH3), 2.35 (s, 3H, Ar–CH3), 3.60–3.65 (m, 2H, H-6′ and H-6′′), 4.14–4.18 (m, 1H, H-5′), 4.34 (dd, 1H, J = 2.1 and 7.8 Hz, H-4′), 4.37 (dd, 1H, J = 2.4 and 5.1 Hz, H-2′), 4.67 (dd, 1H, J = 2.4 and 7.8 Hz, H-3′), 5.60 (d, 1H, J = 5.1 Hz, H-1′), 5.97 (s, 1H, H-3), 6.07 (s, 1H, N–H), 7.20 (d, 2H, J = 8.4 Hz, H-3′′ and H-5′′), 7.40 (td, 1H, J = 1.5 and 8.0 Hz, H-6), 7.42 (d, 2H, J = 8.4 Hz, H-2′′ and H-6′′), 7.48–7.56 (m, 2H, H-5 and H-7), 8.50 (dd, 1H, J = 1.5 and 8.4 Hz, H-8).
13C NMR (75.0 MHz, CDCl3) δ 21.0 (Ar-CH3), 23.7 (CH3), 24.3 (CH3), 25.5 (CH3), 25.7 (CH3), 44.2 (C-6′), 65.1 (C-5′), 70.6 (C-2′), 70.9 (C-3′), 72.0 (C-4′), 96.4 (C-1′), 98.9 (C-3), 108.9 (C-7′), 109.7 (C-8′), 115.9 (C-2′′ and C-6′′), 120.1 (C-5), 122.3 (C-4a), 123.0 (C-8), 125.9 (C-6), 127.5 (C-1), 129.6 (C-7), 130.0 (C-3′′ and C-5′′), 134.2 (C-4′′), 134.7 (C-8a), 140.7 (C-1′′), 153.7 (C-4), 178.1 (C-2).
Anal. Calc. for C29H33N3O6: C, 67.04; H, 6.40; N, 8.09. Found: C, 67.05; H, 6.88; N, 7.73%.
O), 3422 (N–H) and 1588 (CN).
1H NMR (500.00 MHz, CDCl3) δ 1.34 (s, 3H, CH3), 1.39 (s, 3H, CH3), 1.50 (s, 3H, CH3), 1.53 (s, 3H, CH3), 3.60–3.70 (m, 2H, H-6′ and H-6′′), 4.15–4.17 (m, 1H, H-5′), 4.34 (dd, 1H, J = 2.0 and 8.0 Hz, H-4′), 4.37 (dd, 1H, J = 2.5 and 4.5 Hz, H-2′), 4.67 (dd, 1H, J = 2.5 and 8.0 Hz, H-3′), 5.60 (d, 1H, J = 4.5 Hz, H-1′), 6.03 (s, 1H, H-3), 6.33 (m, 1H, N–H), 7.46–7.50 (m, 1H, H-5), 7.46 (d, 2H, J = 8.5 Hz, H-3′′ and H-5′′), 7.36 (d, 2H, J = 8.5 Hz, H-2′′ and H-6′′), 7.40 (td, 1H, J = 1.5 and 7.5 Hz, H-6), 7.55 (td, 1H, J = 1.5 and 7.5 Hz, H-7), 8.44 (dd, 1H, J = 1.5 and 7.5 Hz, H-8), 15.60 (N–N–H).
13C NMR (125.0 MHz, CDCl3) δ 24.2 (CH3), 24.9 (CH3), 26.0 (CH3), 26.1 (CH3), 44.3 (C-6′), 65.0 (C-5′), 70.6 (C-2′), 70.9 (C-3′), 72.1 (C-4′), 96.4 (C-1′), 99.7 (C-3), 108.9 (C-7′), 109.7 (C-8′), 116.6 (C-4′′), 117.2 (C-2′′ and C-6′′), 120.3 (C-5), 122.6 (C-4a), 123.2 (C-8), 126.5 (C-6), 127.9 (C-1), 130.0 (C-7), 132.4 (C-3′′ and C-5′′), 134.4 (C-8a), 142.1 (C-1′′), 154.3 (C-4), 179.9 (C-2).
Anal. Calc. for C28H30BrN3O6: C, 57.54; H, 5.17; N, 7.19. Found: C, 55.78; H, 5.26; N, 6.53%.
O), 3400 (N–H), 1595 (CN) and 2220 (CN).
1H NMR (300.00 MHz, CDCl3) δ 1.35 (s, 3H, CH3), 1.40 (s, 3H, CH3), 1.51 (s, 3H, CH3), 1.54 (s, 3H, CH3), 3.58–3.72 (m, 2H, H-6′ and H-6′′), 4.14–4.17 (m, 1H, H-5′), 4.34 (dd, 1H, J = 1.5 and 7.5 Hz, H-4′), 4.38 (dd, 1H, J = 2.4 and 5.0 Hz, H-2′), 4.68 (dd, 1H, J = 2.4 and 7.5 Hz, H-3′), 5.60 (d, 1H, J = 5.0 Hz, H-1′), 5.92 (s, 1H, H-3), 6.33 (m, 1H, N–H), 7.43 (td, 1H, J = 1.5 and 7.0 Hz, H-6), 7.48–7.55 (m, 1H, H-5), 7.51 (d, 2H, J = 9.0 Hz, H-2′′ and H-6′′), 7.59 (td, 1H, J = 1.5 and 7.0 Hz, H-7), 7.64 (d, 2H, J = 9.0 Hz, H-3′′ and H-5′′), 8.44 (dd, 1H, J = 1.5 and 7.0, Hz, H-8), 15.81 (N–N–H).
13C NMR (75.0 MHz, CDCl3) δ 24.2 (CH3), 24.8 (CH3), 26.0 (CH3), 26.1 (CH3), 44.1 (C-6′), 65.0 (C-5′), 70.5 (C-2′), 70.9 (C-3′), 72.0 (C-4′), 96.4 (C-1′), 98.9 (C-3), 105.5 (CN), 108.9 (C-7′), 109.7 (C-8′), 115.9 (C-2′′ and C-6′′), 119.4 (C-4′′), 120.3 (C-5), 123.4 (C-4a), 123.5 (C-8), 127.2 (C-6), 128.5 (C-1), 129.9 (C-7), 133.6 (C-3′′ and C-5′′), 134.1 (C-8a), 147.7 (C-1′′), 154.1 (C-4), 179.6 (C-2).
Anal. Calc. for C29H30N4O6: C, 65.65; H, 5.70; N, 10.56. Found: C, 65.64; H, 5.83; N, 10.27%.
O), 3340 (N–H), 1595 (CN).
1H NMR (300.00 MHz, CDCl3) δ 1.33 (s, 3H, CH3), 1.51 (s, 3H, CH3), 3.66–3.82 (m, 2H, H-5′ and H-5′′), 4.41 (d, 1H, J = 2.4 Hz, H-3′), 4.45–4.47 (m, 1H, H-4′), 4.63 (d, 1H, J = 3.3 Hz, H-2′), 5.90 (s, 1H, H-3), 6.03 (d, 1H, J = 3.3 Hz, H-1′), 6.40 (m, 1H, N–H), 7.08–7.12 (m, 1H, H-4′′), 7.24-7.27 (m, 1H, H-6), 7.41–7.45 (m, 1H, H-7), 7.35–7.39 (m, 2H, H-3′′ and H-5′′), 7.41–7.45 (m, 2H, H-2′′ and H-6′′), 7.49 (dd, J = 0.9 and 8.0 Hz, H-5), 8.31 (dd, J = 0.9 and 7.8 Hz, H-8), 15.52 (N–N–H).
13C NMR (75.0 MHz, CDCl3) δ 26.1 (CH3), 26.7 (CH3), 42.6 (C-5′), 75.6 (C-3′), 77.5 (C-4′), 85.6 (C-2′), 98.4 (C-3), 104.8 (C-1′), 111.9 (C-6′), 115.8 (C-2′′ and C-6′′), 120.4 (C-5), 122.2 (C-4a), 122.9 (C-8), 124.3 (C-4′′), 126.2 (C-6), 127.6 (C-1), 129.7 (C-7), 129.4 (C-3′′ and C-5′′), 134.4 (C-8a), 142.8 (C-1′′), 154.3 (C-4), 174.4 (C-2).
Anal. Calc. for C24H25N3O5: C, 66.19; H, 5.79; N, 9.65. Found: C, 66.43; H, 5.93; N, 9.29%.
O), 3350 (N–H) and 1592 (CN).
1H NMR (500.00 MHz, DMSO) δ 1.22 (s, 3H, CH3), 1.35 (s, 3H, CH3), 3.47–3.52 (m, 1H, H-5′), 3.60–3.66 (m, 1H, H-5′′), 4.10 (d, 1H, J = 4.0 Hz, H-3′), 4.34–4.40 (m, 1H, H-4′), 4.46 (d, 1H, J = 4.0 Hz, H-2′), 5.75 (s, 1H, H-3), 5.87 (d, 1H, J = 4.0 Hz, H-1′), 7.45–7.48 (m, 1H, H-6), 7.46 (d, 2H, J = 9.0 Hz, H-2′′ and H-6′′), 7.55 (d, 2H, J = 9.0 Hz, H-3′′ and H-5′′), 7.58 (td, J = 0.9 and 7.5 Hz, H-7), 7.90 (dd, 1H, J = 5.5 Hz, N–H), 8.05 (dd, J = 0.9 and 7.5 Hz, H-5), 8.36 (dd, J = 0.9 and 7.5 Hz, H-8), 15.81 (N–N–H).
13C NMR (125.0 MHz, DMSO) δ 26.1 (CH3), 26.7 (CH3), 41.9 (C-5′), 73.7 (C-3′), 77.9 (C-4′), 85.1 (C-2′), 97.6 (C-3), 104.3 (C-1′), 110.7 (C-6′), 115.1 (C-4′′), 117.1 (C-2′′ and C-6′′), 122.4 (C-5), 122.5 (C-8), 123.3 (C-4a), 126.7 (C-6), 128.5 (C-1), 130.0 (C-7), 132.4 (C-3′′ and C-5′′), 133.5 (C-8a), 142.3 (C-1′′), 154.2 (C-4), 178.3 (C-2).
Anal. Calc. for C24H24BrN3O5: C, 56.04; H, 4.70; N, 8.17. Found: C, 56.74; H, 5.02; N, 7.71%.
O), 3365 (N–H), 1592 (CN) and 2224 (CN).
1H NMR (300.00 MHz, DMSO) δ 1.23 (s, 3H, CH3), 1.36 (s, 3H, CH3), 3.63–3.71 (m, 1H, H-5′′), 3.63–3.71 (m, 1H, H-5′), 4.12 (d, 1H, J = 2.7 Hz, H-3′), 4.34–4.39 (m, 1H, H-4′), 4.47 (d, 1H, J = 4.0 Hz, H-2′), 5.77 (s, 1H, H-3), 5.87 (d, 1H, J = 4.0 Hz, H-1′), 7.51 (td, 1H, J = 1.2 and 8.1 Hz, H-6), 7.58–7.64 (m, 1H, H-7), 7.62 (d, 2H, J = 8.7 Hz, H-2′′ and H-6′′), 7.79 (d, 2H, J = 8.7 Hz, H-3′′ and H-5′′), 8.0 (dd, 1H, J = 5.0 Hz, N–H), 8.08 (dd, 1H, J = 1.2 and 8.1 Hz, H-5), 8.40 (dd, 1H, J = 1.2 and 8.1 Hz, H-8).
13C NMR (75.0 MHz, DMSO) δ 26.0 (CH3), 26.7 (CH3), 42.0 (C-5′), 73.6 (C-3′), 77.9 (C-4′), 85.0 (C-2′), 97.6 (C-3), 104.2 (CN), 104.3 (C-1′), 110.6 (C-6′), 115.3 (C-2′′ and C-6′′), 119.4 (C-4′′), 121.8 (C-5), 122.6 (C-8), 123.9 (C-4a), 126.4 (C-6), 130.1 (C-7), 130.3 (C-1), 133.9 (C-3′′ and C-5′′), 134.0 (C-8a), 146.6 (C-1′′), 154.7 (C-4), 178.6 (C-2).
Anal. Calc. for C25H24N4O5: C, 65.21; H, 5.25; N, 12.17. Found: C, 65.19; H, 5.47; N, 11.64%.
X-Ray determination
X-Ray diffraction was performed using an Oxford Diffraction Xcalibur Atlas Gemini ultradiffractometer at 150 K with Mo-Kα (14c) and Cu-Kα (14j) radiation. The collection and reduction of data was performed with CrysalisPro software. The structures (Table 4) were solved by direct methods and refined by full-matrix least squares on F2 with the SHELX-97 software package.49 CCDC reference number 889649 for 14c and 889650 for 14j.| — | 14c | 14j |
|---|---|---|
| Chemical formula | C25H26O5N3Br | C24H24O5N3Br |
| M r | 528.4 | 514.37 |
| Crystal size/mm | 0.39 × 0.14 × 0.07 | 0.25 × 0.15 × 0.03 |
| Crystal color, habit | Purple, needle | Purple, needle |
| Cell setting | Orthorhombic | Orthorhombic |
| Space group | P212121 | P212121 |
| a/Å | 5.8640(2) | 5.1446(3) |
| b/Å | 15.2666(18) | 13.3386(10) |
| c/Å | 26.181(2) | 33.974(3) |
| V/Å3 | 2343.8(4) | 2331.3(3) |
| Z | 4 | 4 |
| μ/mm−1 | 1.80 | 2.75 |
| T min, Tmax | 0.651, 0.894 | 0.664, 1 |
| R int | 0.042 | 0.086 |
| R[F2 > 2σ(F2)], wR(F2), S | 0.029, 0.058, 0.96 | 0.053, 0.133, 1.10 |
| No. reflections | 4782 | 2404 |
| No. parameters | 310 | 316 |
| Δρmax, Δρmin/e Å−3 | 0.43, −0.33 | 0.70, −0.57 |
Cytotoxicity against cancer cell lines
Compounds (0.009–5 μg mL−1) were tested for cytotoxic activity against four cancer cell lines: SF-295 (glioblastoma), HCT-116 (colon), MDA-MB-435 (melanoma), HL-60 (leukemia) (National Cancer Institute, Bethesda, MD). All cell lines were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum, 2 mM glutamine, 100 U/mL penicillin, and 100 μg mL−1 streptomycin at 37 °C with 5% CO2. Each compound was dissolved with DMSO to obtain a stock solution of 1 mg mL−1. The final concentration of DMSO in the culture medium was kept constant below 0.1% (v/v) and the negative control received the same amount of DMSO. Compounds were incubated with cells for 72 h. Doxorubicin (0.18–1.06 μM) was used as a positive control. Cell viability was determined by reduction of the yellow dye 3-(4,5-dimethyl-2-thiazol)-2,5-diphenyl-2H-tetrazolium bromide (MTT) to a blue formazan product as described by Mosmann.50Cell membrane disruption
The test was performed in 96-well plates using a 2% mouse erythrocyte suspension in 0.85% NaCl containing 10 mM CaCl2. The diluted compounds were tested at 200 μg mL−1 as mentioned above. DMSO was used as a negative control and Triton X-100 (1%) was used as positive control. After incubation at room temperature for 1 h followed by centrifugation, the supernatant was removed, the liberated hemoglobin was measured spectrophotometrically at 540 nm and EC50 was calculated. EC50 is the calculated effective concentration that induced 50% of the lysis generated with Triton X-100.Acknowledgements
This work was supported by the Brazilian agency FAPERJ-PRONEX. Fellowships granted to U. F. F., by CAPES, CNPq, CNPq-PIBIC are gratefully acknowledged. We would like to thank the LabCri/UFMG for the use of X-ray facilities.References
- C. M. Galmarini, J. R. Mackey and C. Dumontet, Lancet Oncol., 2002, 3, 415–424 CrossRef CAS.
- W. B. Parker, Chem. Rev., 2009, 109, 2880–2893 CrossRef CAS.
- S. M. Park, H. Yang, S.-K. Park, H. M. Kim and B. H. Kim, Bioorg. Med. Chem. Lett., 2010, 20, 5831–5834 CrossRef CAS.
- P. L. Bonate, L. Arthaud, W. R. Cantrell Jr., K. Stephenson, J. A. Secrist III and S. Weitman, Nat. Rev. Drug Discovery, 2006, 5, 855–863 CrossRef CAS.
- Q. Wanga, X. Liu, Q. Wang, Y. Zhang, J. Jiang, X. Guo, Q. Fan, L. Zheng, X. Yu, N. Wang, Z. Pan, C. Song, W. Qi and J. Chang, Biochem. Pharmacol., 2011, 81, 848–855 CrossRef.
- Y.-S. Lee, S. M. Park, H. M. Kim, S.-K. Park, K. Lee, C. W. Lee and B. H. Kim, Bioorg. Med. Chem. Lett., 2009, 19, 4688–4691 CrossRef CAS.
- M. Aouida, A. Leduc, H. Wang and D. Ramotar, Biochem. J., 2004, 384, 47–58 CrossRef CAS.
- A. K. Choudhury, Z.-F. Tao and S. M. Hecht, Org. Lett., 2001, 3, 1291–1294 CrossRef CAS.
- J.-C. Chapuis, R. M. Schmaltz, K. S. Tsosie, M. Belohlavek and S. M. Hecht, J. Am. Chem. Soc., 2009, 131, 2438–2439 CrossRef CAS.
- M. Facompré, B. Baldeyrou, C. Bailly, F. Anizon, C. Marminon, M. Prudhomme, P. Colson and C. Houssier, Eur. J. Med. Chem., 2002, 37, 925–932 CrossRef.
- Y. Liang, H. Zhou, F. Du, Y.-W. Liu, G.-L. Zou, C.-X. Wang and S.-S. Qu, Thermochim. Acta, 2002, 383, 1–11 CrossRef CAS.
- M. N. da Silva, V. F. Ferreira and M. C. B. V. de Souza, Quim. Nova, 2003, 26, 407–416 CrossRef CAS.
- E. N. S. Júnior, M. C. B. V. de Souza, A. V. Pinto, M. C. F. R. Pinto, M. O. F. Goulart, F. W. A. Barros, C. Pessoa, L. V. Costa-Lotufo, R. C. Montenegro, M. O. de Moraes and V. F. Ferreira, Bioorg. Med. Chem., 2007, 15, 7035–7041 CrossRef.
- C. F. J. Franco, A. K. Jordão, V. F. Ferreira, A. C. Pinto, M. C. B. V. de Souza, J. A. L. C. Resende and A. C. Cunha, J. Braz. Chem. Soc., 2011, 22, 187–193 CAS.
- G. J. Kapadia, M. A. Azuine, V. Balasubramanian and R. Sridhar, Pharmacol. Res., 2001, 43, 363–367 CrossRef CAS.
- V. K. Tandon, H. K. Maurya, N. N. Mishra and P. K. Shukla, Eur. J. Med. Chem., 2009, 44, 3130–3137 CrossRef CAS.
- T. M. S. Silva, C. A. Camara, T. P. Barbosa, A. Z. Soares, L. C. da Cunha, A. C Pinto and M. D. Vargas, Bioorg. Med. Chem., 2005, 13, 193–196 CrossRef CAS.
- V. F. Ferreira, A. Jorqueira, A. M. T. Souza, M. N. da Silva, M. C. B. V. de Souza, R. M. Gouvêa, C. R. Rodrigues, A. V. Pinto, H. C. Castro, D. O. Santos, H. P. Araújo and S. C. Bourguignon, Bioorg. Med. Chem., 2006, 14, 5459–5466 CrossRef CAS.
- R. F. S. Menna-Barreto, D. G. Beghini, A. T. S. Ferreira, A. V. Pinto, S. L. de Castro and J. Perales, J. Proteomics, 2010, 73, 2306–2315 CrossRef CAS.
- P. F. Carneiro, M. C. F. R. Pinto, T. S. Coelho, B. C. Cavalcanti, C. Pessoa, C. A. de Simone, I. K. C. Nunes, N. M. de Oliveira, R. G. de Almeida, A. V. Pinto, K. C. G. de Moura, P. A. da Silva and E. N. S. Júnior, Eur. J. Med. Chem., 2011, 46, 4521–4529 CrossRef CAS.
- S. B. Ferreira, D. T. G. Gonzaga, W. C. Santos, K. G. L. Araújo and V. F. Ferreira, Rev. Virtual Quim., 2010, 2, 140–160 CAS.
- A. S. Prakash, A. G. Moore, V. Murray, C. Matias, W. D. McFadyen and G. Wickham, Chem.-Biol. Interact., 1995, 95, 17–28 CrossRef CAS.
- K. A. Parker and Q.-J. Ding, Tetrahedron, 2000, 56, 10249–10254 CrossRef CAS.
- E. D. Saad, G. Facina and L. H. Gebrim, Rev. Bras. Cancerol., 2007, 53, 47–53 Search PubMed.
- M. Bakker, W. T. A.. Van der Graaf, H. M. J. Groen, E. F. Smith and E. G. E. De Vries, Curr. Pharm. Des., 1995, 1, 133–144 CAS.
- A. Sparreboom, K. Nooter and J. Verweij, The Cancer Handbook, 1st edn, John Wiley & Sons, 2002 Search PubMed.
- G. Minotti, P. Menna, E. Salvatorelli, G. Cairo and L. Gianni, Pharmacol. Rev., 2004, 56, 185–229 CrossRef CAS.
- J. Cummings and J. F. Smyth, Ann. Oncol., 1993, 4, 533–543 CAS.
- G. Capranico, M. Binaschi, M. E. Borgnetto, F. Zunino and M. Palumbo, Trends Pharmacol. Sci., 1997, 18, 323–329 CAS.
- C. Temperini, M. Cirilli, M. Aschi and G. Ughetto, Bioorg. Med. Chem., 2005, 13, 1673–1679 CrossRef CAS.
- Z. S. Saify, N. Mushtaq, F. Noor, S. Takween and M. Arif, Pak. J. Pharm. Sci., 1999, 12, 21–31 CAS.
- J. E. Deweese and N. Osheroff, Nucleic Acids Res., 2008, 37, 738–748 CrossRef.
- A. Rabbani, R. M. Finn and J. Ausió, BioEssays, 2005, 27, 50–56 CrossRef CAS.
- Y. Su, J. Xie, Y. Wang, X. Hu and X. Lin, Eur. J. Med. Chem., 2010, 45, 2713–2718 CrossRef CAS.
- Q. Gong, L. Menon, T. Ilina, L. G. Miller, J. Ahn, M. A. Parniak and R. Ishima, Chem. Biol. Drug Des., 2011, 77, 39–47 CAS.
- A. K. Jordão, P. C. Sathler, V. F. Ferreira, V. R. Campos, M. C. B. V. de Souza, H. C. Castro, A. Lannes, A. Lourenco, C. R. Rodrigues, M. L. Bello, M. C. S. Lourenco, G. S. L. Carvalho, M. C. B. Almeida and A. C. Cunha, Bioorg. Med. Chem., 2011, 19, 5605–5611 CrossRef.
- V. Alagarsamy, V. R. Solomon, M. Murugan, K. Dhanabal, P. Parthiban and G. V. Anjana, J. Enzyme Inhib. Med. Chem., 2008, 23, 839–847 CrossRef CAS.
- A. K. Jordão, V. F. Ferreira, E. S. Lima, M.C. B. V. de Souza, E. C. L. Carlos, H. C. Castro, R. B. Geraldo, C. R. Rodrigues, M. C. B. Almeida and A. C. Cunha, Bioorg. Med. Chem., 2009, 17, 3713–3719 CrossRef.
- S. Rollas and S. G. Küçükgüzel, Molecules, 2007, 12, 1910–1939 CrossRef CAS.
- R. Graeser, N. Esser, H. Unger, I. Fichtner, A. Zhu, C. Unger and F. Kratz, Invest. New Drugs, 2009, 28, 14–19 CrossRef.
- F. Dosio, P. Brusa and L. Cattel, Toxins, 2011, 3, 848–883 CrossRef CAS.
- L. R. Morgan, B. S. Jursic, C. L. Hooper, D. M. Neumann, K. Thangaraja and B. LeBlanc, Bioorg. Med. Chem. Lett., 2002, 12, 3407–3411 CrossRef CAS.
- M. U. Roslund, K. D. Klika, R. L. Lehtilä, P. Tähtinen, R. Sillanpää and R. Leino, J. Org. Chem., 2004, 69, 18–25 CrossRef CAS.
- I. H. Hall, N. J. Peaty, J. R. Henry, J. Easmon, G. Heinisch and G. Pürstinger, Arch. Pharm., 1999, 332, 115–123 CrossRef CAS.
- K. Effenberger-Neidnicht and R. Schobert, Cancer Chemother. Pharmacol., 2010, 67, 867–874 CrossRef.
- S. Rollas and S. G. Küçükgüzel, The Open Drug Delivery Journal, 2008, 2, 77–85 CrossRef CAS.
- R. Mahato, W. Tai and K. Cheng, Adv. Drug Delivery Rev., 2011, 63, 659–670 CrossRef CAS.
- D. R. da Rocha, A. C. G. de Souza, J. A. L. C. Resende, W. C. Santos, E. A. dos Santos, C. Pessoa, M. O. de Moraes, L. V. Costa-Lotufo, R. C. Montenegro and V. F. Ferreira, Org. Biomol. Chem., 2011, 9, 4315–4322 CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 479–485 CrossRef.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS.
Footnote |
| † CCDC reference numbers 889649 and 889650. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra21514d |
| This journal is © The Royal Society of Chemistry 2012 |