Molecular organization and doping in poly(2-methoxyaniline)/Ni(dmit)2 films obtained with the Langmuir–Blodgett technique
Paulo H. S.
Picciani
*a,
Felippe J.
Pavinatto
b,
Nadia M.
Comerlato
c,
Guilherme
Coutinho
ac and
Osvaldo N.
Oliveira
Jr
b
aUniversidade Federal do Rio de Janeiro, Instituto de Macromoléculas (IMA/UFRJ), Av. Horácio de Macedo, 2030, Bloco J, Cidade Universitária, CEP: 21941-598, Rio de Janeiro, RJ, Brazil. E-mail: picciani@ima.ufrj.br; Fax: +55-21-22701317; Tel: +55-21-25628104
bUniversidade de São Paulo, Instituto de Física de São Carlos, Departamento de Física e Ciência dos Materiais, IFSC-USP, Caixa-Postal: 369, Centro, CEP: 13566-590, São Carlos, SP, Brazil. E-mail: pavinatto@ifsc.usp.br; chu@ifsc.usp.br
cUniversidade Federal do Rio de Janeiro, Instituto de Química, Departamento de Química Inorgânica., Av. Athos da Silveira Ramos, 149, Bloco A, 7o. andar, Cidade Universitária, CEP: 21941-909, Rio de Janeiro, RJ, Brazil. E-mail: nadia@iq.ufrj.br
First published on 12th October 2012
Abstract
The control of the properties of materials at the molecular level is pursued for many applications, especially those associated with nanostructures. In this paper, we show that the coordination compound [Ni(dmit)2], where (dmit) is the 1,3-dithiole-2-thione-4,5-dithiolate ligand, can induce doping of poly(2-methoxyaniline) (POMA) in molecularly ordered Langmuir and Langmuir–Blodgett (LB) films. Doping was associated with interactions between the components and the compression of the Langmuir film at the air–water interface, according to polarization-modulated infrared reflection-absorption spectroscopy (PM-IRRAS) data. Taking these results together with in situ UV-Vis absorption measurements, we could identify the molecular groups involved in the interaction, including the way they were reoriented upon film compression. The Langmuir films were sufficiently stable to be transferred as Y-type LB films, while the hybrid POMA/[Ni(dmit)2] films remain doped in the solid state. As expected, the molecular charges affected the film morphology, as observed from combined atomic and electric force microscopy measurements. In summary, with adequate spectroscopy and microscopy tools we characterized molecular-level interactions, which may allow one to design molecular electronic devices with controlled electrical properties.
Introduction
There has been growing interest in using hybrid materials to exploit synergistic effects, especially with compounds possessing distinctive electroactive, optical and magnetic properties. Conducting polymers1 have been important in this context, in particular, polyaniline (PANI) and its derivatives due to their easy synthesis, low cost and environmental stability.2 In conducting polymers, high electrical conductivity can be obtained by doping, either by injecting or removing electrons from the π system of the polymer backbone, or via protonation as in polyanilines.3 A myriad of compounds have been used to dope polyanilines, including protonic acids,4,5 metal ions,6,7 metal particles and nanoparticles,8 metal complexes9 and molecular conductors such as tetracyanoquinodimethane (TCNQ).10The use of the building block 1,3-dithiole-2-thione-4,5-dithiolate (dmit) as a doping agent for polyaniline was proposed earlier,11 and is realized in the present work where attempts were made to investigate doping in controlled supramolecular structures fabricated using the Langmuir–Blodgett (LB) technique.12 The two main features of interest in the LB technique for this specific case are the possibility to organize the molecules at the air–water interface and the synergy of film properties that can be achieved with the molecular-level interactions. Inspiration can be sought, for example, from the superconducting regime obtained with highly organized LB films of [Au(dmit)]2 complexes.13,14 Evidence that a polyaniline derivative could be suitable for the hybrid films is clear from the literature on LB films from conducting polymers used in molecular devices.15 For instance, Ferreira and co-workers reported LB films of PANI and a ruthenium complex for detecting dopamine,16 and Amaya et al. were able to control the conformation of a polyaniline derivative using transition metals due to the strong d–π interactions.17
In this paper, we report on hybrid Langmuir and LB films made with poly(2-methoxyaniline) (POMA) and a [Ni(dmit)2](CTAB)2 complex. POMA was chosen owing to its higher solubility in organic solvents such as chloroform. Using in situ spectroscopy measurements for the Langmuir films at the air–water interface, we show doping of POMA induced by the complex, which depended on the structuring of the molecules. The doped state was preserved when the films were transferred onto solid substrates, in the form of LB films.
Experimental details
POMA synthesis
Poly(2-methoxyaniline) was obtained from the oxidative polymerization of o-anisidine (Sigma-Aldrich, USA) using 1 mol L−1 HCl solution and ammonium persulfate as oxidant. The polymer was obtained in the conducting emeraldine salt form and transformed into emeraldine base (EB) after treatment with 0.1 mol L−1 NH4OH solution for 24 h. The dark blue solid EB obtained was filtered, washed and dried under dynamic vacuum. The spectroscopic characterization with FTIR and UV-Vis absorption confirmed that POMA was obtained. The main IR bands of a KBr pellet with POMA appeared at 1207 cm−1 (stretching of secondary aromatic amine), 1260 and 1029 (C–O–C stretching of alkyl aryl ether linkage), 1460 (C–H bending of the –OCH3 group), 1494 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching of the benzenoid rings) and 1600 (CC stretching of the quinoid rings).19 In the UV-Vis spectrum bands were observed at 330 nm, assigned to the π–π* transition in 1-methyl-2-pyrrolidone, and at 635 nm, assigned to the benzenoid to quinoid transition.11
C stretching of the benzenoid rings) and 1600 (CC stretching of the quinoid rings).19 In the UV-Vis spectrum bands were observed at 330 nm, assigned to the π–π* transition in 1-methyl-2-pyrrolidone, and at 635 nm, assigned to the benzenoid to quinoid transition.11
[Ni(dmit)2](CTAB)2 synthesis
The coordination compound [Ni(dmit)2](CTAB)2 was synthesized following the procedure described in the literature.18 In summary, to a stirred suspension of 4,5-bis(benzoylthio)-1,3-dithiole-2-thione in MeOH, a sodium methoxide solution was added. The resulting deep red solution of Na2(dmit) (0.1 mol) was added under stirring to a solution of NiCl2 in MeOH. After stirring for 30 min at room temperature, a solution of hexadecyltrimethylammonium bromide (CTAB) in MeOH (0.1 mol) was added, from which a dark blue solid precipitated. Elemental anal. calc. for C44H84N2S10Ni: C, 51.78; H, 8.30; N, 2.74. Found C, 51.06; H, 9.11; N, 2.56%. IR (CsI): 2923, 2852 (C–H); 1453 (CC); 1049, 1026 (CS); 914 (C–S); 468, 323 cm−1 (Ni–S).19 The representations of two synthesized compounds are shown in Fig. 1.
2 coordination compound.](/image/article/2012/RA/c2ra21828c/c2ra21828c-f1.gif) | ||
| Fig. 1 Chemical structures of (a) POMA and (b) [Ni(dmit)2](CTAB)2 coordination compound. | ||
Langmuir and Langmuir–Blodgett films
Chloroform solutions of [Ni(dmit)2](CTAB)2, POMA and POMA/[Ni(dmit)2](CTAB)2 mixtures with five molar proportions (indicated in the results section) were prepared at 0.75 mg mL−1. A KSV5000 trough (total volume of 1250 mL) placed in a class 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 clean room was used for surface pressure–area (π–A) isotherm measurements. Solutions were spread over the subphase containing ultrapure water supplied by a Millipore system, with resistivity ≈ 18.2 MΩ cm, pH ≈ 5.5–6.0, surface tension ≈ 72.5 mN m−1. Aliquots of these solutions were spread carefully drop by drop on the surface of the aqueous subphase, after which the solvent was allowed to evaporate over 10–15 min before starting film compression with two movable barriers at a rate of 10 mm min−1. Surface pressure (π) was measured during film compression using a Wilhelmy plate.
000 clean room was used for surface pressure–area (π–A) isotherm measurements. Solutions were spread over the subphase containing ultrapure water supplied by a Millipore system, with resistivity ≈ 18.2 MΩ cm, pH ≈ 5.5–6.0, surface tension ≈ 72.5 mN m−1. Aliquots of these solutions were spread carefully drop by drop on the surface of the aqueous subphase, after which the solvent was allowed to evaporate over 10–15 min before starting film compression with two movable barriers at a rate of 10 mm min−1. Surface pressure (π) was measured during film compression using a Wilhelmy plate.
In situ measurements of electronic and vibrational spectra were performed for Langmuir films at different values of surface pressure. Polarization-modulated infrared reflection-absorption spectra (PM-IRRAS) were obtained with a KSV PMI550 instrument (Biolin Scientific, Helsinki, Finland) in the range from 800 to 4000 cm−1 with a resolution of 8 cm−1. The IR beam was impinged on the water surface with an angle of incidence of ca. 80° being then reflected to the detector. Simultaneous measurements of the spectra for s- and p-polarizations were taken by continuous modulation of the incident light by using a polarizer modulator model PEM100 (Hinds Instruments). The two signals were processed leading to a differential reflectivity spectrum ΔR = (Rp − Rs)/(Rp + Rs), where Rp and Rs are the polarized reflectivity for parallel and perpendicular directions to the plane of incidence, respectively. Further details of the experimental setup can be found in ref. 20. The electronic spectra in the visible region were collected with a portable spectrophotometer model USB2000+ (Ocean Optics, USA), the light source being a LS-1 tungsten halogen lamp, also from Ocean Optics (USA). Plastic optical fibers (50 μm in diameter) were used to guide the light to the surface, and to collect the transmitted light and lead it to the spectrophotometer. The beam crossed the floating monolayer twice being reflected on a mirror placed in the bottom of the trough (inside the subphase) and the absorbed light was estimated by the transmitted light. Full spectra were collected in the region from 400 to 850 nm at different stages of monolayer compression. Further details can be obtained in ref. 21.
Langmuir–Blodgett (LB) films were deposited onto optical glass slides and gold coated optical glass slides. All substrates were previously cleaned with a KOH 5% ethanol solution in an ultrasonic bath for 5 min. The LB transfer was performed with a dipping speed of 2 mm min−1 at a constant surface pressure of 35 mN m−1, with the first layer being deposited during the upstroke. For multilayer LB films, an interval of 10 min elapsed at the most upward position for drying before the subsequent dipping. Film deposition was followed by monitoring the transfer ratio provided by the Langmuir trough. The LB films were characterized by PM-IRRAS, ultraviolet-visible absorbance spectroscopy (UV-Vis) and scanning probe microscopy in atomic force microscopy (AFM) and electric force microscopy (EFM) modes. AFM measurements were performed with OTESPA Si semiconducting probes with a typical force constant of 42 N m−1 and a resonance frequency of 340 kHz using Veeco DI Nanoscope IIIa equipment at the National Center for Research in Energy and Materials (CNPEM), Campinas, Brazil (project number: AFM – 9637). EFM measurements were carried out with the same AFM set up using the two-pass technique, where in the first pass topology is recorded and in the second pass the semiconducting tip follows the recorded topology with a 5 V bias voltage applied between the tip (positively charged) and the grounded sample. As a result, the charged tip is moved according to the charge distribution on the sample, and a charge distribution map can be recorded as a function of tip deflection. Ultraviolet-visible absorbance spectroscopy of the LB films was performed in a Cary 100 spectrophotometer and PM-IRRAS measurements were performed using the same procedures described above for Langmuir films for samples deposited over gold covered glass slides.
Results and discussion
Langmuir films
The surface pressure isotherms of POMA/[Ni(dmit)2](CTAB)2 hybrid films with five molar proportions are shown in Fig. 2. The molar concentration of POMA was calculated based on its repeating unity (r.u.) (∼480 g mol−1).22 The spreading and compression of POMA films onto a pure water subphase led to stable Langmuir films,23,24 whose physicochemical behavior depended on chemical characteristics of the polymer, with the Langmuir–Blodgett transfer being strongly influenced by the molecular mass.24 For the POMA sample used here, the isotherm of the pure polymer (curve 1) has a highly compressible liquid phase that suffers a transition at 3 mN m−1 (indicated by the arrow) to a less compressible liquid-condensed phase. This two-phase isotherm was reported for other POMA systems with molecular weight of 28000 g mol−1.24 Moreover, POMA aggregates were observed on the Langmuir film, which is consistent with its low processability.26 Monolayer collapse took place at ca. 30 mN m−1, identified as a smooth change in the isotherm slope.
2 films in five molar proportions; (1 : 0), (3 : 1), (1 : 1), (1 : 3) and (0 : 1).](/image/article/2012/RA/c2ra21828c/c2ra21828c-f2.gif) | ||
| Fig. 2 Surface pressure versus mean area per repeating unit of POMA/[Ni(dmit)2](CTAB)2 films in five molar proportions; (1:0), (3:1), (1:1), (1:3) and (0:1). | ||
Stable Langmuir monolayers of polyaniline and derivatives can be obtained by decreasing the subphase pH,23,25 and the acid doping induced organization of the polymer chains in Langmuir films.26 Similar behavior was observed here for the hybrid Langmuir films. For POMA/[Ni(dmit)2](CTAB)2 (3:1), three regions corresponding to gas, liquid-expanded and liquid-condensed phases, are seen in curve 2 of Fig. 2. In addition, the collapse pressure increased to ca. 50 mN m−1. As reported in ref. 11, the [In(dmit)2]Cs complex may interact with polyaniline chains and induce an increase in electrical conductivity, similarly to doping with protonic acids. Moreover, the incorporation of the [Ni(dmit)2]2− complex may induce organization of the polymer molecules in the Langmuir films owing to similar interactions to those with acid molecules.22,26 When the proportion of Ni(dmit)2 increased (curves 3 and 4), a sharp transition from the gaseous to the liquid-condensed phase occurred around 45–55 A2/molecule. This behavior can be ascribed to the molecular interaction of Ni(dmit)22− with polymer molecules in a more organized phase. All hybrid POMA/[Ni(dmit)2](CTAB)2 films exhibited lower compressibility than POMA, with collapse pressure between 40–50 mN m−1. These results guided our decision to choose 25, 30 and 35 mN m−1 as suitable pressure values for LB film deposition, since we assumed that all Langmuir films were highly organized at this pressure due to strong interactions between the film components. In fact, such interactions were confirmed by the in situ electronic and vibrational spectroscopy measurements of the Langmuir films, as discussed below. They also affected the extrapolated areas per molecule in Fig. 2. Since POMA is likely to form non-monomolecular films, it is not possible to infer how molecular packing (in terms of occupied areas per molecule) was affected by interaction with [Ni(dmit)2](CTAB)2. It is nevertheless clear that a closer packing could be reached, as one could expect from the strong interactions.
Because hybrid films with the three proportions studied had similar π–A isotherms in Fig. 2, we selected only the 1:1 mixture for further analysis in Langmuir and LB films. Fig. 3 shows the PM-IRRAS spectra27,28 for Langmuir films of (a) POMA, (b) [Ni(dmit)2](CTAB)2 and (c) POMA/[Ni(dmit)2](CTAB)2 1:1, at three surface pressures (0, 10 and 35 mN m−1). In our experimental setup, vertically oriented dipoles are mostly sensitive to the p-polarized (parallel to the plane of incidence) light beam, while horizontally oriented dipoles are sensitive to s-polarized light (perpendicular to the plane of incidence). Thus, the differential spectra are generally surface-specific and provide information on oriented moieties, since molecules in the subphase have random orientations.20
2 and (c) POMA/[Ni(dmit)2](CTAB)2 1 : 1 obtained at surface pressures of 0, 10 and 35 mN m−1.](/image/article/2012/RA/c2ra21828c/c2ra21828c-f3.gif) | ||
| Fig. 3 PM-IRRAS spectra of (a) POMA, (b) [Ni(dmit)2](CTAB)2 and (c) POMA/[Ni(dmit)2](CTAB)2 1:1 obtained at surface pressures of 0, 10 and 35 mN m−1. | ||
Fig. 3(a) shows the characteristic bands at 1020 and 1260 cm−1 for a pure POMA Langmuir film, which are assigned to the symmetric and asymmetric stretching of the alkyl aryl ether linkage, respectively.29,30 Both alkyl aryl ether linkage vibrations present upward bands, which allow us to infer that dipoles for symmetric (μsym) and asymmetric (μasym) transitions are preferentially oriented parallel to the water surface. Furthermore, this orientation is induced by film compression, since the peak is only seen for π above 0 mN m−1. Fig. 3(b) displays increasingly intense, well defined bands at 1025 cm−1, 1062 cm−1 and 1345 cm−1 as the surface pressure increases. The band at 1345 cm−1 can be assigned to the C–N stretching of the CTAB counterion. The doublet at 1025 and 1062 cm−1 appears in (dmit) systems and the higher energy peak (1062 cm−1) is assigned to the stretching vibration of the S2CS terminal segment of the dmit ligand. The lower energy band depends on the molecular symmetry and oxidation state of the ring, in our system being assigned as a Fermi resonance peak between the fundamental state of the stretching vibration of SC and the overtones of S2CS.19 The upward direction in the bands for the [Ni(dmit)2](CTAB)2 film means that such groups are also on the water surface plane. The expected increase in band intensity with the surface pressure indicates not only an increase in surface density but also an orientation of the [Ni(dmit)2](CTAB)2 induced by the film compression.
The spectra for the POMA/[Ni(dmit)2](CTAB)2 (1:1) Langmuir film in Fig. 3(c) depict the same bands assigned to the symmetric and asymmetric stretching of the alkyl aryl ether linkage for the pure POMA monolayer, at 1020 and 1260 cm−1. Interestingly, the mixture with [Ni(dmit)2](CTAB)2 caused both bands to be negative for the film in the gaseous phase (π = 0 mN m−1), with the asymmetric band being more pronounced, indicating that POMA ether dipoles for the mixed film in the gaseous phase are mostly oriented upwards. Nevertheless, after the start of film compression, when the surface pressure increased to 10 mN m−1, a drastic molecular reorganization occurred. At this point the system behaved like a liquid phase with molecular areas around 40 A2 mol−1, in such a way that the mobility of the molecules allowed the dipoles of ether linkage bonds to adopt the same conformation as in the pure film, i.e. parallel to the water surface. It seems that the presence of [Ni(dmit)2](CTAB)2 on the surface, and strongly interacting with POMA (as previously observed in π–A isotherms), caused a distinct orientation of the bonds in the gaseous phase compared to the pure monolayer. However, this was surpassed by the competitive effect from film compression; when the surface pressure reached 30 mN m−1 (molecular area is around 30 A2 mol−1), the ether bonds were forced to adopt the horizontal conformation.
An additional band at 1140 cm−1 in Fig. 3(c) is believed to be the same vibration due to the stretching of CS terminal groups (shown in Fig. 3(b)), which is now broadened and shifted to higher energies/wavenumbers (from 1057 to 1140 cm−1). Such modifications of band shape and position support the hypothesis that [Ni(dmit)2](CTAB)2 and POMA interact strongly in the Langmuir film, with the terminal CS groups of the complex taking part. In addition, the same trend for the 1260 cm−1 vibration of POMA was observed for the CS groups, with reorientation from the gaseous to more packed states. The band was oriented upward at low pressures, which means that CS groups were located on the water surface plane, as in the pure [Ni(dmit)2](CTAB)2 film. After film compression, the band was oriented downward with the CS groups adopting a vertical orientation. For the mixed film this band was less defined and less intense than for the pure [Ni(dmit)2](CTAB)2 monolayer, which suggests that CS groups are on average less oriented in the presence of POMA.
The proposed orientation for [Ni(dmit)2](CTAB)2 in pure monolayers and for POMA/[Ni(dmit)2](CTAB)2 Langmuir films is illustrated in Scheme 1 for different surface pressures. Of major relevance was the reorientation undergone by both materials in the mixed film when the monolayer changed from the gaseous to a more compact phase (π > 0 mN m−1). Furthermore, [Ni(dmit)2](CTAB)2 adopted an orientation that differed from the one in the pure film, and CS terminal groups interacted strongly with POMA, as inferred from the broadening and shift in the 1057 cm−1 band.
2 orientation in the pure monolayer (left) and [Ni(dmit)2](CTAB)2 and POMA arrangement in the mixed 1 : 1 Langmuir monolayer for different surface pressures.](/image/article/2012/RA/c2ra21828c/c2ra21828c-s1.gif) | ||
| Scheme 1 Illustration of the proposed [Ni(dmit)2](CTAB)2 orientation in the pure monolayer (left) and [Ni(dmit)2](CTAB)2 and POMA arrangement in the mixed 1:1 Langmuir monolayer for different surface pressures. | ||
Further information on the molecular arrangement of the film components can be obtained from the electronic absorption spectra obtained in situ for the Langmuir films, shown in Fig. 4.
2, and (c) POMA/[Ni(dmit)2](CTAB)2 at distinct π values.](/image/article/2012/RA/c2ra21828c/c2ra21828c-f4.gif) | ||
| Fig. 4 UV-Vis absorption spectra for Langmuir films of (a) POMA, (b) [Ni(dmit)2](CTAB)2, and (c) POMA/[Ni(dmit)2](CTAB)2 at distinct π values. | ||
In its emeraldine base form, POMA presented no detectable signal for the monolayer in the gaseous phase or in the packed state at 40 mN m−1 (Fig. 4(a)). In contrast, the [Ni(dmit)2](CTAB)2 complex showed increasing signal with compression for two regions, between 410 and 460 nm and above 600 nm, as shown in Fig. 4(b). The shape of the spectrum can be analyzed by comparing with the UV-Vis absorption spectra for these materials in chloroform solutions, shown in Fig. 5. The broad band between 410 and 460 nm in the Langmuir film corresponds to that centered at 400 nm for the solution spectrum, which is assigned to the π(Sm)–π*(CS) transition.31 The other part of the spectra that slightly increases in intensity with film compression (above 600 nm) is difficult to analyze. It is probable that the band around 560 nm in the solution spectrum (assigned to metal–ligand charge transfer transitions31) is superimposed with weak, broad bands for [Ni(dmit)2](CTAB)2 around 650 and 770 nm due to π(Sm)–π*(CS) or metal–ligand charge transfer transitions, respectively.31 This difficulty arises because the in situ UV-Vis spectra for Langmuir films normally have a less defined shape, with band shifts caused by aggregation.21
2 and POMA/[Ni(dmit)2](CTAB)2 in chloroform solutions.](/image/article/2012/RA/c2ra21828c/c2ra21828c-f5.gif) | ||
| Fig. 5 UV-Vis absorption spectra for POMA, [Ni(dmit)2](CTAB)2 and POMA/[Ni(dmit)2](CTAB)2 in chloroform solutions. | ||
For the hybrid POMA/[Ni(dmit)2](CTAB)2 Langmuir film, small absorptions at shorter wavelengths (<450 nm) are observed in Fig. 4(c) due to [Ni(dmit)2](CTAB)2. A broad absorption band at longer wavelengths (>600 nm) increased with film compression in a much more pronounced fashion than the band for the pure [Ni(dmit)2](CTAB)2 film. Moreover, this band has the shape of the well-known polaronic transition band in polyanilines,25,32 which means that electron delocalization processes may occur in the POMA/[Ni(dmit)2](CTAB)2 system. Therefore, POMA may reach the semiconducting or metallic regime upon interacting with [Ni(dmit)2](CTAB)2, which is consistent with the finding that M(dmit) can induce doping in polyanilines.11 The doping process is not related to the counterions; instead, it can be associated with the oxidation of Ni(dmit)2− anions to Ni(dmit)+ that can add charges to the polymer chains.33 However, it is worth noting that this charge transfer takes place only in highly organized molecules; the polaronic band was not observed for POMA/[Ni(dmit)2](CTAB)2 in solution (Fig. 5). Hence, the doping process in POMA seems to be induced by the formation of Langmuir films. The possible control in film packing is also relevant, since the band was broader under compression. Similar behavior was observed by Zhang and co-workers for a polyaniline–dodecyl benzene sulfonic acid (DBSA) monolayer on a 0.03 M HCl subphase, with the insulator–metal transition being observed upon increasing the surface pressure, especially above 30 mN m−1.34
LB film deposition
The Langmuir–Blodgett films were transferred onto cleaned gold coated glass substrates at 20, 30 and 35 mN m−1. Fig. 6(a) shows non-homogeneous LB films deposited at 25 mN m−1, with the transfer ratio (TR) varying significantly from one deposited layer to the other. More regular TR behavior is observed for deposition at 30 mN m−1 and increased deposition rates were reached at 35 mN m−1. Fig. 6(b) shows the transfer ratios versus layer number for the deposition of pure POMA, [Ni(dmit)2](CTAB)2, and hybrid POMA/[Ni(dmit)2](CTAB)2 LB films deposited at 35 mN m−1. Both films containing Ni(dmit)2 were Y-type in the beginning of the deposition, with TR values converging to the ideal value of 1.0. Similar behavior was reported for POMA layers doped with protonic acids.23,25 For neat POMA, deposition is less regular, but nevertheless the data indicate that some material was deposited on the substrate.2 LB films at 25, 30 and 35 mN m−1 and (b) POMA, [Ni(dmit)2](CTAB)2 and POMA/[Ni(dmit)2](CTAB)2 LB films onto Au coated glass slides.](/image/article/2012/RA/c2ra21828c/c2ra21828c-f6.gif) | ||
| Fig. 6 Transfer ratio versus number of layers for (a) POMA/[Ni(dmit)2](CTAB)2 LB films at 25, 30 and 35 mN m−1 and (b) POMA, [Ni(dmit)2](CTAB)2 and POMA/[Ni(dmit)2](CTAB)2 LB films onto Au coated glass slides. | ||
LB films with 11 layers were further characterized by PM-IRRAS, UV-Vis spectroscopy and scanning probe microscopy. According to the surface selection rules,35 only vibrational modes with dipole moments normal to the surface are IR active.27,28Fig. 7 shows the PM-IRRAS spectra for POMA, [Ni(dmit)2](CTAB)2 and POMA/[Ni(dmit)2](CTAB)2 LB films deposited on Au covered glass slides. Characteristic vibrational bands for POMA at 1530 cm−1 and 1650 cm−1 assigned to benzenoid and quinoid units appear in both POMA and POMA/[Ni(dmit)2](CTAB)2 films.11 Also, a broad, intense band was observed in the POMA Langmuir film at 1280 cm−1, and was assigned to the C–O–C stretching. The spectrum for the [Ni(dmit)2](CTAB)2 LB film exhibits the CS stretching vibration at 1068 cm−1, and therefore this group is oriented perpendicularly to the substrate surface. The POMA/[Ni(dmit)2](CTAB)2 LB film displays the bands of both POMA and [Ni(dmit)2](CTAB)2, consistent with the finding that both components were transferred to the LB film.
2 and POMA/[Ni(dmit)2](CTAB)2 and POMA Langmuir–Blodgett films deposited on Au covered glass slides.](/image/article/2012/RA/c2ra21828c/c2ra21828c-f7.gif) | ||
| Fig. 7 PM-IRRAS spectra of [Ni(dmit)2](CTAB)2 and POMA/[Ni(dmit)2](CTAB)2 and POMA Langmuir–Blodgett films deposited on Au covered glass slides. | ||
The electronic absorption spectra of LB films deposited onto glass slides in Fig. 8 show the expected bands at 320 nm (as a shoulder) and 560 nm for POMA, corresponding to π–π* and benzenoid to quinoid (excitonic) transitions, respectively.11 The [Ni(dmit)2](CTAB)2 LB film spectrum shows bands at 320, 355, 410 and 462 nm due to the intraligand π–π* transition, and at 640 nm due to the metal–ligand charge transfer transition (MLCT).31 All bands are redshifted in comparison to the spectra in solution and two more bands are present due to the existence of intermolecular interactions in the solid state as already observed in [Ni(dmit)2]−2 LB films.36,37 The tail above 1000 nm for the [Ni(dmit)2](CTAB)2 LB film can be compared to the band at 1150 which is assigned to electronic transitions within the [Ni(dmit)]−2 moieties in similar systems.36,37 The hybrid LB film displayed a broad band at 640 nm and a shoulder around 590 nm, assigned to both Ni(dmit)2 and POMA, respectively, and a polaronic band tail (λ > 700 nm) normally observed in doped polyanilines due to π–polaron transitions. The appearance of this tail indicates that the POMA conducting state due to electron delocalization induced by [Ni(dmit)2](CTAB)2 is preserved after the LB film transfer.
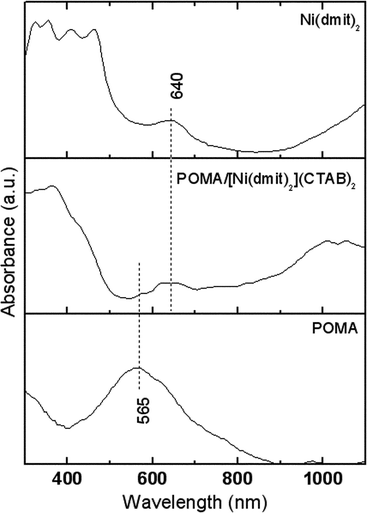 | ||
| Fig. 8 Electronic absorption spectra of Langmuir–Blodgett films deposited on glass slides. | ||
Fig. 9 shows topological AFM images compared to the electric potential contrast obtained at the same time using EFM. The [Ni(dmit)2](CTAB)2 LB film in Fig. 9(a) presents a well defined structure with growth perpendicular to the substrate movement direction (X direction). It seems, however, that a continuous film was not formed, which led to an estimated surface roughness of 184.2 nm for a 1 μm2 area. There is also a strong correlation between the topography and the EFM image, as can be seen in Fig. 9(b). This means that the charge distribution follows the morphology with the peaks in the topography image corresponding to the highly charged parts of the film. For the hybrid POMA/[Ni(dmit)2](CTAB)2 in Fig. 9(c), there are two distinct regions of the sample. One is fiber-like, corresponding to the highly charged parts of the film (see Fig. 9(d)). The other seems to be a granular topography with lower charge density. The fibrous bundles were noted to be perpendicular to the dipping direction. These fiber-like bundle structures have also been found by Aoki and co-workers in a PANI–DBSA system,38 and were attributed to the elongation forces in spreading a PANI solution in chloroform over the water subphase and to the strong π–π interchain interactions. The RMS roughness estimated for hybrid POMA/[Ni(dmit)2](CTAB)2 was 101.8 nm for a 1 μm2 analyzed area, which shows that the addition of POMA significantly increases film homogeneity compared to the Ni(dmit) film. Furthermore, charge distribution on the hybrid films follows the fiber orientation, indicating that the hybrid POMA/[Ni(dmit)2](CTAB)2 structures have a high density of negative charges, possibly due to the doping process over the fibril structures (Fig. 9(d)). The POMA LB film exhibits a granular morphology with no fiber-like structures (Fig. 9(e)), with a RMS roughness of 43.70 nm for a 1 μm2 area. No charge distribution was detected using the EFM technique because POMA was deposited in its insulating, uncharged emeraldine base form.
2, (c) POMA/[Ni(dmit)2](CTAB)2, (e) POMA; and EFM images of (b) [Ni(dmit)2](CTAB)2, (d) POMA/[Ni(dmit)2](CTAB)2, (f) POMA LB films deposited at 35 mN m−1.](/image/article/2012/RA/c2ra21828c/c2ra21828c-f9.gif) | ||
| Fig. 9 AFM images of (a) [Ni(dmit)2](CTAB)2, (c) POMA/[Ni(dmit)2](CTAB)2, (e) POMA; and EFM images of (b) [Ni(dmit)2](CTAB)2, (d) POMA/[Ni(dmit)2](CTAB)2, (f) POMA LB films deposited at 35 mN m−1. | ||
Fig. 10 compares the AFM and EFM images of [Ni(dmit)2](CTAB)2 LB films deposited on Au covered glass slides at 35 and 30 mN m−1. One notes in Fig. 10(a) for the LB film deposited at 35 mN m−1 a highly oriented fiber-like pattern perpendicular to the dipping direction (substrates were moved along the X direction), while Fig. 10(b) shows a granular, non-regular pattern for the film transferred at 30 mN m−1. Furthermore, the EFM images reveal higher charge density along the fibers, indicating that delocalization takes place for LB films deposited at 35 mN m−1 but not for those deposited at 30 mN m−1. Even though the hybrid Langmuir film displayed a well-defined polaronic band at pressures above 10 nM m−1 (Fig. 4(b)), the molecular organization and packing are also important for the electrical properties of the LB film, since charge delocalization only occurred for the films transferred at pressures above 30 mN m−1.
2 hybrid LB films deposited at surface pressures of (a) 35 mN m−1 and (b) 30 mN m−1. The pictures in the insert are EFM images of the corresponding AFM images.](/image/article/2012/RA/c2ra21828c/c2ra21828c-f10.gif) | ||
| Fig. 10 AFM images of [Ni(dmit)2](CTAB)2 hybrid LB films deposited at surface pressures of (a) 35 mN m−1 and (b) 30 mN m−1. The pictures in the insert are EFM images of the corresponding AFM images. | ||
Conclusions
This work provides qualitative experimental findings which show that POMA can be doped by [Ni(dmit)2](CTAB)2 complexes when the molecules are packed in a Langmuir film. Using in situ PM-IRRAS spectroscopy, we could infer that [Ni(dmit)2](CTAB)2 molecules were reoriented upon film compression owing to interaction with the POMA polymer chains, while the dipoles in the POMA ether linkages also changed their orientation. The resulting effect of this new molecular architecture on the electronic transitions of the hybrid film could be probed by in situ electronic spectroscopy in the visible range. POMA molecules behave as a doped conducting polymer, since a well-defined polaronic band was observed with the increase in surface pressure. Since [Ni(dmit)2](CTAB)2 has no acidic species able to promote acid doping in POMA, we can argue that the molecular organization at the air–water interface can mediate the transfer of charges to the polymer backbone leading to a doped POMA as observed using in situ UV-Vis spectroscopy.This new electronic state of the hybrid film persisted in the solid state. Y-type 11-layer LB films could be deposited for the pure and hybrid materials, and the absorption spectra indicated that the hybrid POMA/[Ni(dmit)2](CTAB)2 remained doped after transfer onto the substrate. Also, bundle-like structures were observed by AFM all over the film surface and EFM measurements revealed that the electrical charge density is concentrated along oriented fiber structures, indicating an anisotropic conductive behavior.
Acknowledgements
This work was supported by CNPq, CAPES and FAPESP (Brazil).References
- (a) C. K. Chiang, Y. W. Park, A. J. Heeger, H. E. Shirakawa, J. Louis and A. G. MacDiarmid, J. Chem. Phys., 1978, 69, 5098 CrossRef CAS.
- R. Skotheim, R. L. Elsenbaumer and J. R. Reynolds, Handbook of Conducting Polymers, Marcel Dekker, New York, 2nd edn, 1998, pp 197–409 Search PubMed.
- A. Ray, G. E. Asturias, D. L. Kershner, A. F. Richter, A. G. MacDiarmid and A. J. Epstein, Synth. Met., 1989, 29, 141 CrossRef.
- J-C. Chiang and A. G. MacDiarmid, Synth. Met., 1986, 13, 193 CrossRef CAS.
- M. Reghu, Y. Cao, D. Moses and A. J. Heeger, Synth. Met., 1993, 57, 5020 CrossRef CAS.
- J. Zhang, H. Wang, S. Yang, S. Wang and S. Yang, J. Appl. Polym. Sci., 2012, 125, 2494 CrossRef CAS.
- O. P. Dimitriev, Macromolecules, 2004, 37, 3388 CrossRef CAS.
- S. Tian, J. Liu, T. Zhu and W. Knoll, Chem. Commun., 2003, 2738 RSC.
- O. P. Dimitriev, P. S. Smertenkoa, B. Stillerb and L. Brehmerb, Synth. Met., 2005, 149, 187 CrossRef CAS.
- J. C. Li, Z. Q. Xue, ϒ Y. Zeng, W. M. Liu, Q. D. Wu, Y. L. Song and L. Jiang, Thin Solid Films, 2000, 374, 59 CrossRef CAS.
- P. H. S. Picciani, F. G. Souza JR., N. M. Comerlato and B. G. Soares, Synth. Met., 2007, 157, 1074 CrossRef CAS.
- T. Bjørnholm, T. Hassenkam and N. Reitzel, J. Mater. Chem., 1999, 9, 1975 RSC.
- J.-P. Savy, D. de Caro, L. Valade, J.-P. Legros, P. Auban-Senzier, C. R. Pasquier, J. Fraxedas and F. Senocq, EPL, 2007, 78, 37005 CrossRef.
- Y. F. Miura, M. Horikiri, S.-H. Saito and M. Sugi, Solid State Commun., 2000, 113, 603 CrossRef CAS.
- T-W. Zeng, C-C. Ho, Y-C. Tu, G-Y. Tu, L-Y. Wang and W-F. Su, Langmuir, 2011, 27, 15255 CrossRef CAS.
- M. Ferreira, K. Wohnrath and O. N. Oliveira Jr., Synth. Met., 2003, 135–136, 455 CrossRef CAS.
- T. Amaya, D. Saio, S. Koga and T. Hirao, Macromolecules, 2010, 43, 1175 CrossRef CAS.
- B. M. F. Avila, N. M. Comerlato, R. A. Howie and J. L. Wardell, Inorg. Chim. Acta, 2004, 357, 1487 CrossRef CAS.
- G. Liu, Q. Fang, W. Xu, H. Chen and C. Wang, Spectrochim. Acta, Part A, 2004, 60, 541 CrossRef.
- F. J. Pavinatto, C. Pascholatti, E. A. Montanha, L. Caseli, H. S. Silva, P. B. Miranda, T. Viitala and O. N. Oliveira Jr., Langmuir, 2009, 25, 10051 CrossRef CAS.
- D. S. dos Santos Jr., F. J. Pavinatto, D. T. Balogh, L. Misoguti, O. N. Oliveira Jr. and C. R. Mendonça, J. Colloid Interface Sci., 2004, 276, 138 CrossRef CAS.
- S. Paddeu, M. K. Ram and C. Nicolini, J. Phys. Chem. B, 1997, 101, 4759 CrossRef CAS.
- A. Riul Jr., L. H. C. Mattoso, S. V. Mello, G. D. Telles and O. N. Oliveira Jr., Synth. Met., 1995, 71, 2067 CrossRef.
- R. M. Faria, L. H. C. Mattoso, M. Ferreira, O. N. Oliveira Jr., D. Gonçalves and O. S. Bulhões, Thin Solid Films, 1992, 221, 5 CrossRef CAS.
- M. J. Ram, S. Carrara, S. Paddeu, E. Maccioni and C. Nicolini, Langmuir, 1997, 13, 2760 CrossRef CAS.
- A. Aoki, R. Umehara and K. Banba, Langmuir, 2009, 25, 1169 CrossRef CAS.
- A. Viinikanoja, S. Areva, N. Kocharova, T. Ääritalo, M. Vuorinen, A. Savunen, J. Kankare and J. Lukkari, Langmuir, 2006, 22, 6078 CrossRef CAS.
- R. G. Greenler, J. Chem. Phys., 1966, 44, 310 CrossRef CAS.
- L. Zhang, H. Peng, J. Sui, C. Soeller, P. A. Kilmartin and J. Travas-Sejdic, J. Phys. Chem. C, 2009, 113, 9128 CAS.
- J. Jiang, L.-H. Ai and A.-H. Liu, Synth. Met., 2010, 160, 333 CrossRef CAS.
- C. Arantes, A. M. Rocco and M. L. Rocco, J. Mol. Struct., 2010, 969, 220 CrossRef CAS.
- L. Premvardhana, L. A. Peteanua, P.-C. Wang and A. G. MacDiarmid, Synth. Met., 2001, 116, 157 CrossRef.
- P. Cassoux, Coord. Chem. Rev., 1999, 185–186, 213 CrossRef CAS.
- J. Zhang, A. L. Barker, D. Mandler and P. R. Unwin, J. Am. Chem. Soc., 2003, 125, 9312 CrossRef CAS.
- T. Buffeteau, B. Desbat and J. M. Turlet, Appl. Spectrosc., 1991, 45, 380 CrossRef CAS.
- C. Pearson, A. S. Dhindsa, L. M. Goldenberg, R. A. Singh, R. Dieing, A. J. Moore, M. R. Bryce and M. C. Petty, J. Mater. Chem., 1995, 5(10), 1601 RSC.
- L. M. Goldenberg, C. Pearson, M. R. Bryce and M. C. Petty, J. Mater. Chem., 1996, 6, 699 RSC.
- A. Aoki, R. Umehara and K. Banba, Langmuir, 2009, 25, 1169 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |