Thermal polycondensation of poly(diol citrate)s with tethered quaternary ammonium biocides†
Peter N.
Coneski
,
Preston A.
Fulmer
and
James H.
Wynne
*
Chemistry Division, Code 6100, Naval Research Laboratory, 4555 Overlook Avenue SW, Washington, DC 20375, USA. E-mail: james.wynne@nrl.navy.mil; Fax: +1 (202) 767-0594; Tel: +1 (202) 404-4010
First published on 23rd October 2012
Abstract
The synthesis and characterization of a series of novel, antimicrobial, aliphatic polyesters based on citric acid, alkanediols, and quaternary ammonium salt (QAS) diols is described. These materials possess a wide range of mechanical and thermal properties (storage modulus, E′ = 1.03–17.53 MPa at 25 °C; Tg = −15.7–38.6 °C) primarily dictated by alkanediol selection and the presence of biocide. Surface analysis via contact angle and FTIR-ATR studies indicate the presence of biocide at the material/air interface providing high antimicrobial activity (5–7 log kill of S. aureus over controls) for all materials containing tethered QAS. Hydrolysis of these cross-linked materials was also highly controlled based on the QAS, alkanediol structure, and curing duration; and significant hydrolysis occurs in as little as 4 weeks.
Introduction
The persistence of plastics in the environment has motivated researchers to develop new materials capable of hydrolytic degradation into environmentally benign products in an effort to reduce mounting consumer waste.1 Although numerous synthetic polymers may be classified as degradable, polyesters remain at the forefront of innovation due to their ease of synthesis and diverse mechanical and degradation properties.2–4 Known for their potential as medical materials, such as tissue engineering scaffolds and absorbable implants, degradable polyesters have also been investigated for applications such as degradable packaging materials as an alternative to the current persistent materials (e.g. poly(styrene) (PS), poly(ethylene) (PE), and poly(ethylene terephthalate) (PET)).1Poly(α-esters) (e.g. poly(lactic acid) (PLA) and poly(glycolic acid) (PGA)) and poly(hydroxyalkanoates) have been investigated as specialty materials for medical applications; however, their use remains limited due to the high costs associated with their preparation.1 Poly(diol citrate)s have recently been reported to be a unique alternative to these materials with both diverse mechanical characteristics and facile, inexpensive preparations.5–7 Unfortunately, the lack of any inherent antimicrobial character allows the polymer to harbor bacteria which could lead to the spread of infection, especially throughout a hospital setting.8 While certain methods to impart antibacterial character into inexpensive polyesters have been reported, such as the introduction of nitric oxide releasing functionalities and the dispersion of antimicrobials within the polymer matrix, the relatively short antibacterial window and rapid leaching of biocides from the polymer matrix, respectively, may be problematic.7–9 As such, the covalent immobilization of biocidal moieties onto polyester scaffolds is an important advancement toward the development of antimicrobial materials.
Progress in polymer synthesis methodologies and the preparation of functionalized polymers has brought to light numerous methods for the preparation of functionalized polyesters.10–12 The contributions of “click” functionalized polyester materials has led to the development of numerous drug-conjugated polymers, however the efficient eradication of microbes at the material interface is limited to polymers with pendant biocides capable of disrupting the cellular membrane of bacteria.8,11 Of these, quaternary ammonium salts (QAS) are perhaps the most popular and their tethering to numerous different polymer systems, including polyesters, has been reported.8,13–16 Despite this, rigorous synthetic techniques and high costs limit the potential implementation of “click-able” materials in various high-throughput applications.
Herein, we describe the preparation of antimicrobial polyesters via the thermal polycondensation of citric acid with quaternary ammonium salt (QAS) diols and alkanediols for potential uses as degradable, antimicrobial packaging materials. The influence of starting material composition, reaction stoichiometry, curing temperatures and curing times are investigated as a function of both thermal properties and antibacterial capabilities and related to future utility as environmentally friendly, antibacterial, packaging materials
Experimental section
Materials
All chemicals were purchased from Sigma-Aldrich (Milwaukee, WI) and used as received, unless otherwise noted. Citric acid, 1,8-octanediol, and all common laboratory salts and solvents were purchased from Fisher Scientific. Staphylococcus aureus (ATCC 25923) was used for initial bacterial challenges. Luria-Bertani (LB) broth (Difco Laboratories, Detroit, MI) was used as a bacterial growth medium for preparation of bacteria for all challenges. Letheen broth (Difco Laboratories, Detroit, MI) was used as a dilution media post-challenge due to its ability to deactivate quaternary ammonium salts (QAS).General procedure for the synthesis of quaternary ammonium salt (QAS) diols
Quaternary ammonium salt diols were synthesized according to a previously reported method (Scheme 1).13 Briefly, N-methyldiethanolamine (1.20 mol), bromoalkane (1.20 mol), and ethanol (400 mL) were stirred under reflux for 24 h. After cooling to room temperature, the ethanol was removed in vacuo, and the white residue was redissolved in acetone. The solution was then chilled using a dry ice/acetone bath with vigorous stirring to initiate crystallization of the product. The solid white crystals were then filtered and dried in vacuo overnight.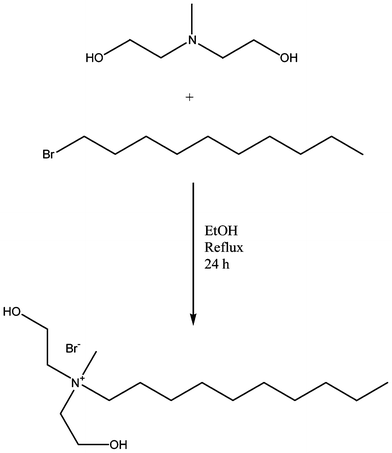 | ||
| Scheme 1 Representative synthesis of Q10 quaternary ammonium salt (QAS) diol. | ||
Butyl bis(2-hydroxyethyl) methyl ammonium bromide (Q4): 1H NMR (D2O, δ): 4.56 (m, 2H), 3.87 (m, 4H), 3.48–3.39 (t, 4H), 3.30–3.25 (t, 2H), 3.00 (s, 3H), 1.65–1.54 (m, 2H), 1.25–1.18 (m, 2H), 0.81–0.76 (t, 3H). 13C NMR (D2O, δ): 63.5, 55.3, 49.7, 23.9, 19.3, 13.2. Yield: 93%
Hexyl bis(2-hydroxyethyl) methyl ammonium bromide (Q6): 1H NMR (D2O, δ): 4.56 (m, 2H), 3.80 (m, 4H), 3.36–3.32 (t, 4H), 3.23–3.17 (t, 2H), 2.94 (s, 3H), 1.54 (m, 2H), 1.10–1.07 (m, 6H), 0.66–0.62 (t, 3H). 13C NMR (D2O, δ): 63.4, 55.2, 49.5, 30.4, 25.2, 21.7, 13.3. Yield: 91%
Octyl bis(2-hydroxyethyl) methyl ammonium bromide (Q8): 1H NMR (CDCl3, δ): 4.80 (m, 2H), 4.10 (m, 4H), 3.75–3.72 (t, 4H), 3.58–3.52 (m, 2H), 3.33 (s, 3H), 1.76 (m, 2H) 1.35–1.27 (m, 10H), 0.90–0.85 (t, 3H). 13C NMR (CDCl3, δ): 64.2, 55.7, 50.5, 31.7, 29.2, 26.4, 22.6, 14.1. Yield: 85%
Decyl bis(2-hydroxyethyl) methyl ammonium bromide (Q10): 1H NMR (CDCl3, δ): 4.80 (m, 2H), 4.11 (m, 4H), 3.77–3.71 (m, 4H), 3.57–3.53 (m, 2H), 3.33 (s, 3H), 1.75 (m, 2H), 1.35–1.26 (m, 14H), 0.90–0.86 (t, 3H). 13C NMR (CDCl3, δ): 64.2, 55.7, 50.5, 31.8, 29.4, 29.2, 26.4, 22.6, 14.1. Yield: 92%
General procedure for the synthesis of poly(diol citrate) elastomers
Poly(diol citrate) elastomers were synthesized via the thermal curing of a prepolymer melt. Formation of the prepolymer melts was accomplished by heating the desired diol mixture (0.15 total mol diol) with citric acid (0.10 mol) at 155 °C (Scheme 2). After completely melting, the solution continued to stir for 5 min, at which point it was carefully poured into a mold and cured for 4 or 10 d at elevated temperatures. After curing for the intended time period, the polymers were cooled to room temperature, removed from the mold, and stored in a dry environment at room temperature until analysis.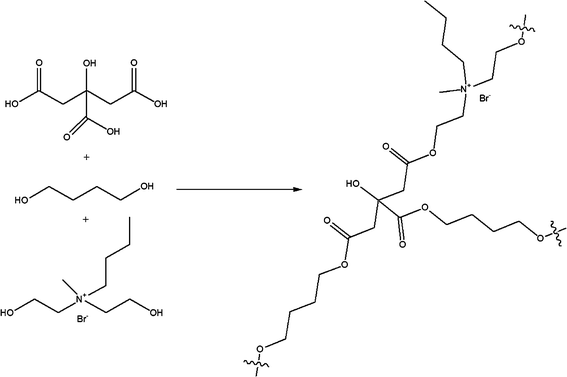 | ||
| Scheme 2 Representative synthesis of poly(diol citrate) elastomers prepared from 1,4-butanediol and Q4. | ||
Characterization
Polymer decomposition temperatures were analyzed using a TA Instruments Q50 TGA using heating rates of 10 °C min−1 in an N2 atmosphere. Glass transition temperatures were measured using a TA Instruments Q20 DSC, with heating and cooling rates of 10 °C min−1. Glass transition temperatures were identified as the inflection point of the endotherm. Dynamic mechanical analysis was performed using a Perkin Elmer DMA 8000 operated at 1 Hz over a temperature range of −70 °C to 120 °C and a ramp rate of 2 °C min−1. Glass transition temperatures were identified as the peak of the tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ vs. temperature plot. An AST Products VCA Optima goniometer was used to measure contact angles. Gel fractions were measured by soaking polymer samples in THF for 24 h at room temperature. After removal of the solvent, samples were dried overnight to remove residual THF and the gel fraction was calculated as:
δ vs. temperature plot. An AST Products VCA Optima goniometer was used to measure contact angles. Gel fractions were measured by soaking polymer samples in THF for 24 h at room temperature. After removal of the solvent, samples were dried overnight to remove residual THF and the gel fraction was calculated as: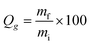 | (1) |
Water uptake and in vitro hydrolysis
Water uptake was assessed by placing elastomer squares of approximately 250 mg into 20 mL of distilled water at room temperature. After 24 h, the swollen polymers were removed from the solution, gently blotted dry, and the mass was recorded. Water uptake was calculated according to the following equation: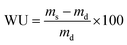 | (2) |
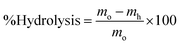 | (3) |
Bactericidal efficacy of poly(diol citrate)s
Due to its prevalence in hospital settings, the antibacterial activity of the poly(diol citrate)s synthesized was measured against Staphylococcus aureus. S. aureus was grown at 37 °C until reaching log phase growth. Bacterial cells were harvested by centrifugation, counted on a hemocytometer using phase contrast microscopy, pelleted by centrifugation at 4000 g for 10 min, and resuspended in PBS at a concentration of 1 × 109 CFU mL−1. To prevent dessication of the bacteria during antimicrobial testing, a hydration chamber was prepared using a sterile 3 × 3 in gauze pad placed in the bottom of a sterile 150 × 15 mm Petri dish. Test samples were placed on the gauze pad, which was saturated with 5 mL of sterile water. An aliquot of the bacterial suspension (10 μL) containing 1 × 107 bacteria was added to each test polymer, and placed in the hydration chamber at room temperature. After incubation for 2 h, the bacteria were recovered by placing the sample in a tube containing 5 mL sterile Letheen media, followed by 30 s of vortexing. After serial dilutions were carried out, the solutions were incubated at 37 °C with agitation. After 18 h, the cultures were examined for turbidity, which indicated bacterial growth. Each coating was tested in triplicate, with the log kill determined by the following: log kill = 7 − highest dilution exhibiting bacterial growth. All bacterial challenge procedures were conducted using standard aseptic techniques in a BSL-2 hood.Results and discussion
The formulation of cross-linked polyesters from the thermal polycondensation of polyols with polyacids has a number of advantages including simple synthetic techniques that do not require the use of potentially toxic catalysts or crosslinking agents and excellent control over the resulting material properties.9,17 Furthermore, a number of metabolites, such as glycerol and citric acid, and other environmentally friendly precursors, have been polymerized in such a manner to form non-toxic degradable materials.5–7,9 As a result, these types of polymers have exhibited great promise as new degradable materials for biomedical and packaging applications.1,3 Unfortunately, many of these types of materials lack any inherent antibacterial character and are prone to colonization by a variety of pathogenic microbes that can transmit disease to those that come into contact with the material, especially for packaging materials of medical supplies. This study sought to examine the synthesis and resulting material and antibacterial properties of cross-linked aliphatic polyesters that contain covalently bound quaternary ammonium salt (QAS) diols within the polymer chain to impart a defense mechanism against microbial contamination. Despite previous reports detailing mechanical property modulation of cross-linked polyester matrices via strategic control over curing temperatures, the incorporation of the thermally sensitive QAS diols prohibited extreme variations in curing temperatures in this investigation.17,18 Specifically, curing of prepolymers containing even small amounts of the biocidal diols at temperatures over 100 °C resulted in the formation of discolored non-uniform materials, often times with low gel fractions likely resulting from the thermal decomposition of the QAS moieties (data not shown). As a result, curing the prepolymers at 80 °C, which resulted in the formation of optically transparent, uniform polymers with no discoloration, was deemed optimal. Additionally, the 80 °C curing temperatures utilized in this study allowed for the utilization of citric acid to diol ratios of 2:3, despite previous reports indicating solid network formation using 1:1 reaction stoichiometry.5 Within these parameters, 37 different poly(diol citrate) compositions were prepared and cured for both 4 and 10 d, resulting in the preparation of 74 different material formulations (Table 1, ESI†).
| Polymer | Diol(s) | Cure time (d) |
|---|---|---|
| PEC-a/b | 100% Ethylene glycol | a – 4, b – 10 |
| PBC-a/b | 100% Butanediol | a – 4, b – 10 |
| PHC-a/b | 100% Hexanediol | a – 4, b – 10 |
| POC-a/b | 100% Octanediol | a – 4, b – 10 |
| PDC a/b | 100% Decanediol | a – 4, b – 10 |
| PEC-Q10-a/b | 75% Ethylene glycol:25% Q10 | a – 4, b – 10 |
| PBC-Q10-a/b | 75% Butanediol:25% Q10 | a – 4, b – 10 |
| PHC-Q4-a/b | 75% Hexanediol:25% Q4 | a – 4, b – 10 |
| PHC-Q6-a/b | 75% Hexanediol:25% Q6 | a – 4, b – 10 |
| PHC-Q8-a/b | 75% Hexanediol:25% Q8 | a – 4, b – 10 |
| PHC-Q10-a/b | 75% Hexanediol:25% Q10 | a – 4, b – 10 |
| POC-Q10-a/b | 75% Octanediol:25% Q10 | a – 4, b – 10 |
| PDC-Q4-a/b | 75% Decanediol:25% Q4 | a – 4, b – 10 |
| PDC-Q6-a/b | 75% Decanediol:25% Q6 | a – 4, b – 10 |
| PDC-Q8-a/b | 75% Decanediol:25% Q8 | a – 4, b – 10 |
| PDC-Q10-a/b | 75% Decanediol:25% Q10 | a – 4, b – 10 |
In general, it was discovered that the degree of polymerization of poly(diol citrate)s was greatly influenced by the alkanediol and QAS diol structure, as evidenced by the gel fraction of cured materials (Table 2). As the alkanediol substituent became larger, the degree of crosslinking increased similarly, as indicated by the gel fraction trends which show that POC-a (highest gel fraction) ≥ PDC-a > PHC-a > PBC-a > PEC-a (lowest gel fraction). Curing of prepolymer melts containing diol content of 100% ethylene glycol resulted in the formation of completely soluble low-molecular-weight byproducts as opposed to the highly cross-linked matrix intended, possibly occurring from the loss of ethylene glycol during thermal curing which would result in an excess of acid monomer and subsequent lower degrees of reaction. Differences in gel fraction likely occur as a result of the increased number of conformational states of the alkanediols of greater size (e.g. 1,10-decanediol) in the melt phase which enhanced their rate of crosslinking over those diols whose conformations remain limited (e.g. ethylene glycol). Despite this, longer curing times are able to offset evidence of this trend as all alkanediols copolymerized with citric acid showed appreciable crosslinking (greater than 97% gel fraction) at 10 d of curing with the exception of PEC-b. In general, replacement of even a portion of the alkanediol components inhibited polymerization of the poly(diol citrate)s, as all QAS diol-containing formulations remained tacky and fluid after 4 d of curing and only those containing 25% QAS diol and 75% alkanediol fully cured even after 10 d (ESI†). Decreased cross-linking of networks containing QAS may occur for a variety of reasons including inaccessibility and/or deactivation of reactive groups. Specifically, inaccessibility of reactive groups may be caused by the collapse of the long alkyl chain onto free hydroxyl or carboxyl functionalities preventing them from undergoing further reaction. Deactivation of functional groups is a more likely occurrence and may occur as a result of tertiary amines (a byproduct of the thermal decomposition of quaternary ammonium salts) deprotonating the carboxylic acid groups of citric acid forming resonance stabilized carboxylate anions, which cannot be esterified in the presence of free hydroxyl groups. Although decomposition of the quaternary ammonium moiety was not observed acutely at 80 °C, prolonged exposure to high temperatures may cause sufficient decomposition to inhibit complete crosslinking of the polyester matrix. Curing beyond 4 d likely provides sufficient reaction time to impart cross-linking to the materials, despite inactivation of some carboxylic acid functionalities.
| Sample | Gel fraction (%) | Decomposition temperature/°C | Glass transition temperature/°C |
|---|---|---|---|
| PEC-a | 0.0 ± 0.0 | 253 | 8.4 |
| PEC-b | 15.70 ± 1.75 | 257 | 38.6 |
| PBC-a | 83.91 ± 0.62 | 274 | 3.1 |
| PBC-b | 100.29 ± 0.98 | 277 | 24.6 |
| PHC-a | 88.97 ± 0.32 | 310 | −10.3 |
| PHC-b | 97.43 ± 0.65 | 307 | −0.5 |
| POC-a | 91.60 ± 2.31 | 308 | −16.7 |
| POC-b | 99.94 ± 1.09 | 334 | −11.2 |
| PDC-a | 89.79 ± 1.81 | 319 | −17.1 |
| PDC-b | 100.32 ± 0.63 | 335 | −15.7 |
| PHC-Q4-b | 96.85 ± 0.56 | 239 | 8.1 |
| PHC-Q6-b | 97.76 ± 0.06 | 236 | 10.7 |
| PHC-Q8-b | 97.03 ± 0.33 | 235 | 8.7 |
| PHC-Q10-b | 95.21 ± 2.57 | 225 | 2.6 |
| POC-Q10-b | 94.98 ± 0.59 | 236 | 0.3 |
| PDC-Q4-b | 98.10 ± 0.38 | 232 | −2.2 |
| PDC-Q6-b | 96.39 ± 0.21 | 235 | −5.5 |
| PDC-Q8-b | 97.27 ± 0.41 | 240 | −2.4 |
| PDC-Q10-b | 96.25 ± 0.31 | 228 | −1.4 |
As QAS diols have been shown to be thermally sensitive, ensuring that the incorporation of these biocides occurs without excessive decomposition is a crucial aspect of the successful preparation of a variety of antimicrobial packaging materials.19 FTIR-ATR spectra indicate that indeed a large portion of the QAS species remain during the curing procedures. Definitive C–N stretching attributed to quaternary ammonium species is present at 1634 cm−1 for materials containing QAS, but is absent for poly(diol citrate)s that do not contain the quaternary ammonium functionality (Fig. 1).20 These results also confirm that biocides are present at the surface of the material, which is a decisive element contributing to the biocidal behavior of these materials.
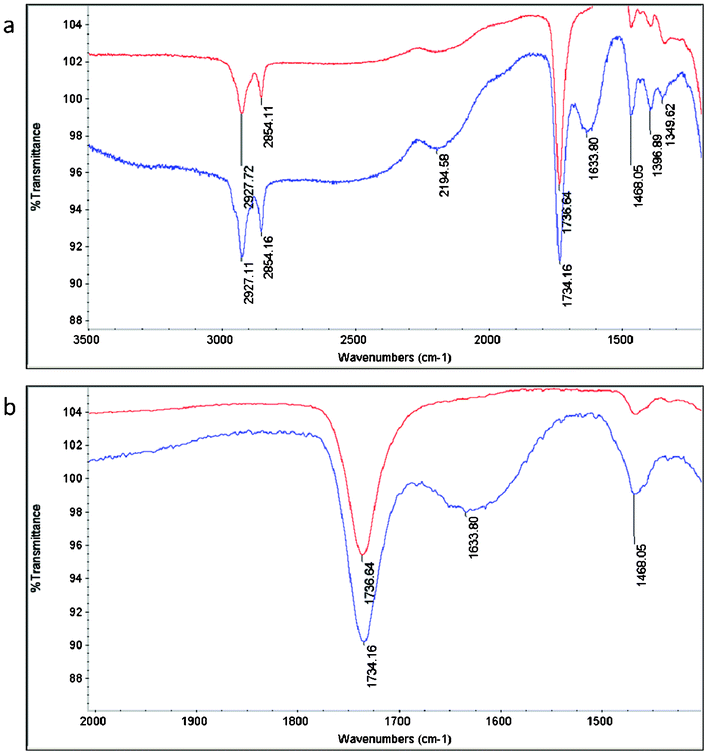 | ||
| Fig. 1 (a) Full and (b) expanded FTIR-ATR spectra of PDC-b (red line) and PDC-Q10-b (blue line) indicating incorporation of QAS as indicated by the emergence of a characteristic C–N stretching pattern at 1633.8 cm−1. Note: peak intensities were purposely offset to increase peak clarity. | ||
In addition to controlling esterification rates of the polyester scaffolds, alkanediol selection is perhaps more importantly an integral component in controlling the thermal characteristics of the resulting polymers. Thermal transitions are particularly important characteristics for packaging materials as they can be used to elucidate the flexibility of materials within specific temperature ranges. In this study, as the size of the alkanediols increased, the glass transition temperature (Tg) of the cured elastomers decreased sequentially from 38.6 °C for PEC-b to −15.7 °C for PDC-b, an expected trend based on the decreased crosslink density associated with the incorporation of longer alkanediol components (Table 2, Fig. 2). Additionally, longer curing times resulted in the formation of a larger number of matrix-stabilizing crosslinks and subsequent higher Tg values. Tethered QAS diols along the polymer backbone resulted in an overall increase in the glass transition temperatures for the cured materials caused by decreased chain mobility arising from the addition of bulky substituents along the polymer chains. Values increased by 3–12 °C upon addition of QAS diols to hexanediol copolymers while those for decanediol increased by 10–15 °C. Interestingly, there were no observable Tg trends based on the length of the alkyl segment of the QAS.
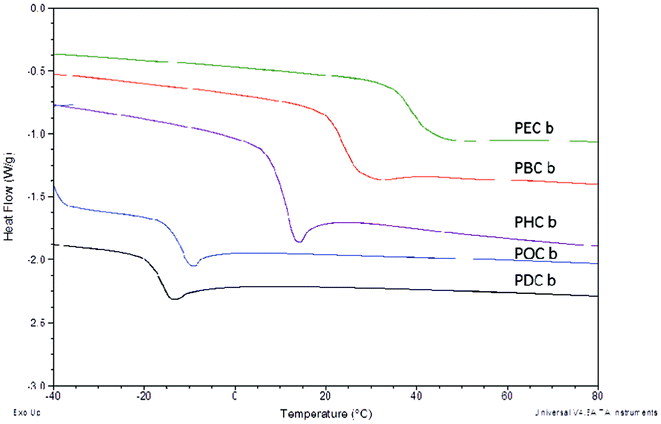 | ||
| Fig. 2 DSC thermograms of poly(diol citrate) elastomers showing correlation of alkanediol structure on resulting glass transition temperatures. DSC was performed using a TA Instruments Q20 DSC, with heating and cooling rates of 10 °C min−1. Glass transition temperatures were identified as the inflection point of the endotherm. | ||
Glass transition temperatures acquired using dynamic mechanical analysis were in close agreement with those obtained using DSC, as the largest deviation between analysis techniques was only 8.3 °C (Fig. 3). Furthermore, aside from PBC-b, all Tg values obtained by DMA were within 2.9 to 8.3 °C higher than those acquired using DSC. As expected based on glass transition temperatures, the storage moduli of the poly(diol citrate)s synthesized decreased with increasing alkanediol chain length from 533.6 MPa for PBC-b to 5.02 MPa for PDC-b at 0 °C and from 17.53 MPa to 5.46 MPa at 25 °C (Table 3). Comparable to DSC analysis, DMA also showed increased Tg values for QAS tethered polymers compared to their parent poly(diol citrate)s without quaternized substituents. These increased Tg values also manifested themselves in increased moduli at 0 °C compared to the unquaternized counterparts, albeit superficially as their parent polymers had already passed through their thermal transition regions. Above the glass transitions for all of the copolymers, the QAS-modified poly(diol citrate)s exhibited moduli lower than those of their respective unmodified materials caused by the interruption of matrix interactions by the pseudo-grafted structures (Table 3). Overall, the materials produced with tethered QAS showed flexibility at room temperature, indicating they may be potential replacements for soft packaging materials in which antimicrobial characteristics would be a benefit.
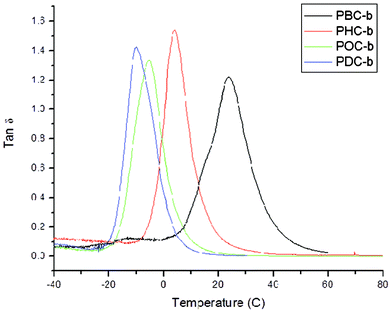 | ||
| Fig. 3 tanδ vs. temperature plot for poly(diol citrate)s showing increased glass transition temperature with increasing alkanediol chain length. DMA was performed using a Perkin Elmer DMA 8000 operated at 1 Hz over a temperature ranges of −70 °C to 120 °C and a ramp rate of 2 °C min−1. Glass transition temperatures were identified as the peak of the tanδ vs. temperature plot. | ||
| Sample | Glass transition temperature/°C | Storage modulus at 0 °C | Storage modulus at 25 °C |
|---|---|---|---|
| PBC-b | 23.7 | 533.6 MPa | 17.53 MPa |
| PHC-b | 3.9 | 168.7 MPa | 6.72 MPa |
| PHC-Q4-b | 14.9 | 1230 MPa | 6.33 MPa |
| PHC-Q6-b | 13.6 | 1100 MPa | 3.86 MPa |
| PHC-Q8-b | 12.3 | 940.5 MPa | 4.01 MPa |
| PHC-Q10-b | 6.8 | 223.8 MPa | 2.11 MPa |
| POC-b | −5.5 | 7.94 MPa | 6.11 MPa |
| POC-Q10-b | 7.4 | 81.8 MPa | 2.62 MPa |
| PDC-b | −9.8 | 5.02 MPa | 5.46 MPa |
| PDC-Q4-b | 2.2 | 53.3 MPa | 3.63 MPa |
| PDC-Q6-b | 2.8 | 59.64 MPa | 2.70 MPa |
| PDC-Q8-b | 1.5 | 45.0 MPa | 2.98 MPa |
| PDC-Q10-b | 4.2 | 47.2 MPa | 1.03 MPa |
As packaging materials are often exposed to a wide range of temperatures, ensuring that thermal decomposition does not occur at normally encountered temperatures is an extremely important factor in determining the utility of such materials. In this study, the onset on thermal decomposition of the polyesters without tethered QAS was directly related to the alkanediol length, as the incorporation of higher molecular weight diols resulted in higher values for decomposition onset (Table 2). As would be expected based on increased monomer consumption and gel fraction, 10 d curing times caused thermal decomposition temperatures to rise slightly compared to compositionally similar materials cured for only 4 d. Upon incorporation of the QAS diols into the matrix, the onset of decomposition dropped drastically due to the well-known thermal decomposition of the biocidal moiety.19 Although the onset of decomposition did not significantly deviate upon increasing QAS content, complete thermal decomposition was achieved at lower temperatures with increasing QAS content, confirming the presence of QAS within the polymer matrix (Fig. 4). Despite this, the overall decomposition temperatures were sufficiently high as to not be a general concern for applications in which these materials are intended.
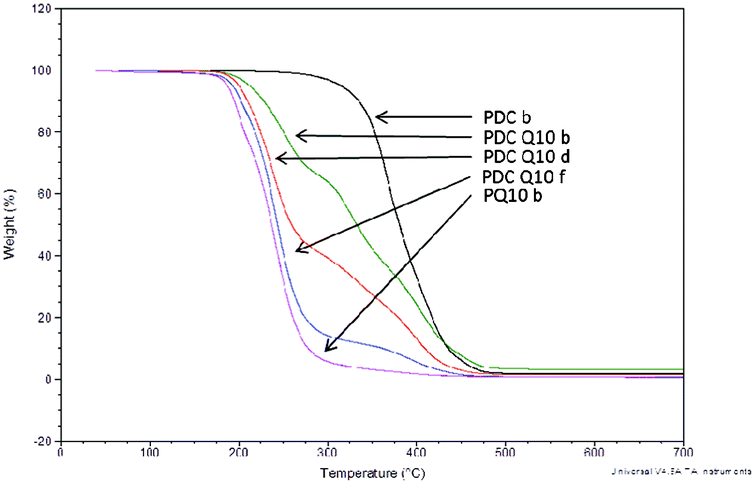 | ||
| Fig. 4 Thermal decomposition onset of poly(diol citrate)s with tethered QAS displaying influence of QAS content on bulk polymer degradation. TGA was performed in an N2 atmosphere using heating rates of 10 °C min−1. | ||
As the surface chemistry of materials containing QAS biocides has previously been shown to be an important factor in their antimicrobial activity, static water contact angle was measured across all cured matrices in order to compare the levels of hydrophobicity and/or hydrophilicity at their surfaces. Despite there being limitations of accuracy of contact angle measurements for hydrolytically unstable materials due to solvent/surface interactions, hydrolysis of these cross-linked polymers was observed to occur sufficiently slowly so that general trends of the surface wetting behavior of the poly(diol citrate)s could be elucidated. The alkyl content of the poly(diol citrate)s made a significant contribution to the overall contact angle of the cured materials as those synthesized with longer chain alkanediols (e.g. decanediol) exhibited higher contact angles than those with shorter diol species (e.g. butanediol) (Fig. 5). Curing time also played a significant role in the resulting contact angle of the materials. POC-a and PDC-a resins, which were both cured for 4 d, possessed significantly lower contact angles than their counterparts that were cured for 10 d (14.3° and 17.9° lower respectively). This is likely a result of the increased polarity/hydrophilicity of carboxylic acid and alcohol end-groups of the less highly cross-linked materials (i.e. POC-a and PDC-a) compared to the ester groups within the polymer chain of their more highly cross-linked counterparts (POC-b and PDC-b). Interestingly, polymers containing 1,4-butanediol and 1,6-hexanediol behaved in an opposite fashion, with those cured for longer time periods possessing slightly lower contact angles than their counterparts, which were cured for shorter time periods. Despite this inconsistency, the complexity of reactive spreading of carboxylic acid-containing surfaces has been reported previously and further examination of this behavior is beyond the scope of this investigation.21 As was expected, copolymerization of QAS diols into the polymer matrix also had a substantial effect on the contact angle of the resulting materials. Incorporation of the hydrophilic QAS moiety resulted in decreased contact angle for all formulations investigated compared to their parent polymers, however the extent of this decrease varied between copolymer systems and QAS structure. For example, QAS copolymerized into decanediol-based poly(diol citrate)s resulted in a uniform decrease for observed contact angles with no dependence on the QAS structure. However, copolymerization into hexanediol-containing systems resulted in more significant contact angle depressions with increasing QAS alkyl tail length. This behavior indicates that the longer alkyl tails of the QAS biocides likely exhibit increased solubility within the bulk polymer matrix as compared to the shorter tails, leaving the hydrophilic quaternary ammonium functionality more available to contribute to the surface hydrophilicity. For materials with shorter tail QAS, the increased alkyl content at the material interface likely contributes some hydrophobic character to balance the hydrophilicity of the quaternary ammonium species, resulting in a more balanced surface chemistry and higher relative contact angle (Fig. 5).
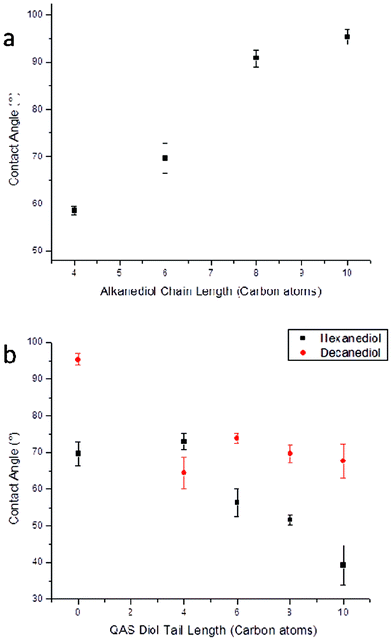 | ||
| Fig. 5 Static water contact angle of poly(diol citrate)s (a) without and (b) with QAS performed using a AST Products VCA Optima goniometer. | ||
Due to the relatively short distance between crosslinking groups and the inability for subsequent phase separation, water uptake of the bulk polymer followed expected trends based on component structure, again despite the polymer/water interactions that are known to occur for hydrolytically unstable materials. Increased alkyl content of the larger alkanediol components resulted in diminished water uptake by the bulk material as a result of increasing material hydrophobicity. Specifically, PBC-b was able to absorb 10.4% of its weight in water after 1 d of soaking at room temperature whereas the more hydrophobic material PDC-b only absorbed 1.3% (Fig. 6a). The increased hydrophilicity of the materials that contained covalently bound QAS was evident by the substantial increases in water uptake values compared to non-QAS samples. For example, upon replacement of 25% of the hexanediol of PHC-b with Q4, the 2.5% water uptake rose to 22.2%. As materials containing QAS with larger alkyl tails have additional hydrophobic contributions from the alkyl segments, associated water uptake was also found to diminish with increasing alkyl chain length, with the trend PDC-Q4-b (highest water uptake) > PDC-Q6-b > PDC-Q8-b > PDC-Q10-b > PDC-b (lowest water uptake) (Fig. 6b). As the main goals of any type of packaging materials are to protect the contents enclosed within, polymers with lower water uptake are more ideal candidates as replacement materials than those with higher degrees of water uptake. This makes the compositions containing longer chain QAS and longer alkanediols more promising than those composed of the shorter chain derivatives.
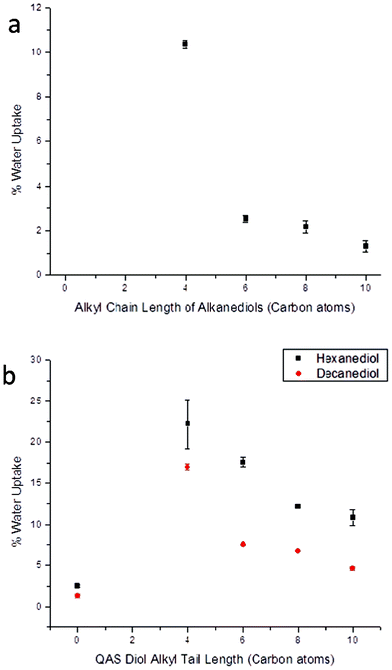 | ||
| Fig. 6 Water uptake of poly(diol citrate)s (a) without and (b) with tethered biocides after 24 h of submersion in distilled water at 25 °C. | ||
One of the characteristics of cross-linked aliphatic polyesters that make them particularly promising as medical and packaging materials is their controlled hydrolysis. Numerous reports have identified various factors influencing the rates of hydrolysis including the overall crosslink density, monomer structure, and hydrophobicity.3,6,17 In this investigation, all of these factors played an important role in the overall hydrolysis of the resulting matrices. The greater hydrophilicity of short chain alkanediols resulted in much more rapid rates of hydrolysis compared to poly(diol citrate)s with long chain alkanediols (Fig. 7a). For instance, PEC-b was completely hydrolyzed within a few weeks of exposure to water whereas POC-b and PDC-b suffered only 2.96% and 1.52% hydrolysis, respectively, after 16 weeks submerged in water. Poly(diol citrate) curing times also had a significant effect on rates of hydrolysis. Increased cross-link formation associated with longer curing times generally resulted in slower hydrolysis rates due to a combination of an increased number of bonds required to be hydrolyzed before an observed mass loss and the increased hydrophobicity for some formulations. Specifically, PBC-a suffered 84.95% mass loss over 16 weeks, while PBC-b, which was cured for over twice as long, only experienced a 50.29% mass loss during the same time period. Likewise, PHC, POC, and PDC samples cured for 4 d time periods endured approximately 3–4× more mass loss over the course of 16 weeks than did their counterparts cured for 10 d. As would be expected for materials with increased hydrophilicity and greater water uptake, incorporation of QAS diols into the poly(diol citrate) structures resulted in increased hydrolysis rates compared to the parent polyesters alone. The replacement of 25% of hexanediol substituents with Q10 in PHC-Q10-b resulted in 25.64% mass loss after 16 weeks compared to only 4.36% mass loss for PHC-b. QAS structure was also found to have a significant effect on the rates of hydrolysis as incorporation of biocides with shorter alkyl chains into the polymer structure resulted in increased hydrolysis compared to those with longer alkyl substituents (Fig. 7b). For example, hydrolysis of PHC-Q4-b for 16 weeks resulted in 35.59% mass loss, while PHC-Q10-b only suffered a 25.64% reduction in the same amount of time. These trends held true for all conjugates as overall hydrolysis of biocide-containing matrices followed the pattern Q4 (fastest hydrolysis) > Q6 > Q8 > Q10 (slowest hydrolysis). Interestingly, all biocide-containing conjugates experienced an accelerated initial mass loss at the first time point compared to the remainder of the study. Although this is partially attributed to extraction of non-cross-linked soluble materials from the matrix, the magnitude of mass loss is greater than that of the total soluble materials within the matrix. This additional mass loss is attributed to increased hydrolysis rates of QAS diols compared to the alkanediol substituents in the polymer. The increased hydrophilicity provided by the ionic diols likely accelerates hydrolysis at proximal ester linkages compared to esters near the more hydrophobic alkanediols. As these substituents hydrolyze and are extracted from the sample matrix, the rates of hydrolysis slow due to the increased hydrophobicity of the remaining material and lower concentration of cross-links near the more hydrophilic QAS.
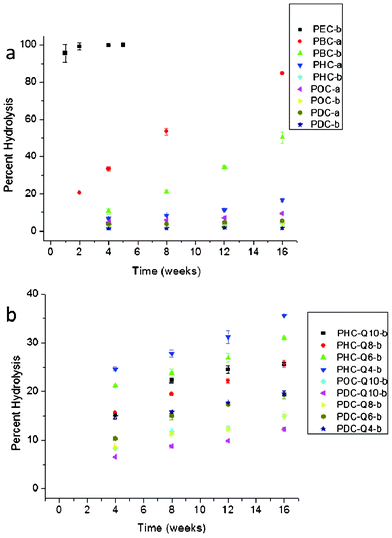 | ||
| Fig. 7 Hydrolysis of poly(diol citrate)s (a) without and (b) with tethered QAS. Hydrolysis was measured as the average mass lost of 3 samples after soaking in distilled water at 25 °C for the intended time periods. Note the differences in scale on the y-axes. | ||
Overall, poly(diol citrate)s with tethered QAS possessed very promising antibacterial characteristics compared to control materials. All polymers containing QAS exhibited a 5–7 log reduction in viable bacteria compared to control materials synthesized without biocide (Fig. 8). Interestingly, there was no observable change in bacterial killing based on the structure of the biocide despite previous reports indicating greater degrees of killing for longer chain alkyl quaternary ammoniums. This behavior may be attributed to the relative density of QAS within the material. As the biocide incorporation of these polymers is based on a mole percent rather than a weight percent, the surface density of QAS is likely higher for lower molecular weight QAS species (i.e.Q4 and Q6) compared to higher molecular weight biocides (Q10). This heightened presence of biocide may be acting as an equalizing factor in the overall performance of these polymeric materials. Evidence of this trend is further supported by the slight decrease in antimicrobial activity for PDC-Q10-b, whose QAS content is the lowest by mass of any of the formulations investigated due to the high relative molar masses of 1,10-decanediol and Q10 compared to other poly(diol citrate) starting materials utilized. Furthermore, the accelerated hydrolysis of QAS-containing materials may be enhancing biocidal activity by acting as a matrix with the controlled release of biocide that is triggered by water uptake. Regardless of mechanism, as many microbial strains are capable of utilizing the hydrolysis products of aliphatic polyesters as nutrient sources, the composition of these scaffolds may promote the initial association of bacteria at the material interface, thereby enhancing the effectiveness of the tethered biocides in killing those that attempt to colonize at the surface over other polymers containing tethered QAS.22–24 Overall, the high degree of antibacterial activity coupled with the unique mechanical and surface properties support further research into the utility of these polymers for a variety of antibacterial applications including medical packaging materials. Investigations are currently under way to fully characterize rates of poly(diol citrate) hydrolysis with respect to monomer content and determine if the ester functionalities promote the adhesion and association of bacteria, thereby increasing the effectiveness of these materials.
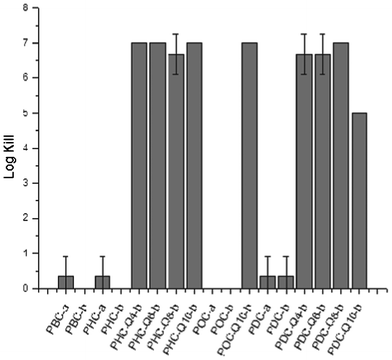 | ||
| Fig. 8 Antimicrobial activity of poly(diol citrate)s against Staphylococcus aureus after 2 h incubation on the polymer matrices. | ||
Conclusions
Degradable poly(diol citrate) polymers with high antibacterial activity were achieved via the covalent incorporation of QAS diols into a crosslinked matrix containing citric acid and alkanediols. These materials had tailorable surface and thermal properties determined by the diol structure and degree of biocide incorporation. Room temperature hydrolysis rates were shown to be influenced by the degree of crosslinking, hydrophobicity of the alkanediol substituents, and structure of QAS biocides, with significant hydrolysis occurring in as little as 4 weeks for PEC-b. Overall, elastomers containing QAS exhibited a 5–7 log reduction in viable S. aureus compared to control materials with bactericidal efficacy independent of QAS structure, making them strong candidates for use in hydrolysable packaging materials for healthcare applications. Further research aimed at material diversification, in-depth hydrolysis product quantification, and more rigorous bacterial challenges is planned.Acknowledgements
This work was funded by the Office of Naval Research and the Naval Research Laboratory.References
- W. Amass, A. Amass and B. Tighe, A Review of Biodegradable Polymers: Uses, Current Developments in the Synthesis and Characterization of Biodegradable Polyesters, Blends of Biodegradable Polymers and Recent Advances in Biodegradation Studies, Polym. Int., 1998, 47, 89–144 CrossRef CAS.
- L. S. Nair and C. T. Laurencin, Biodegradable Polymers as Biomaterials, Prog. Polym. Sci., 2007, 32, 762–798 CrossRef CAS.
- B. Amsden, Curable, Biodegradable Elastomers: Emerging Biomaterials for Drug Delivery and Tissue Engineering, Soft Matter, 2007, 3, 1335–1348 RSC.
- U. Edlund and A.-C. Albertsson, Polyesters Based on Diacid Monomers, Adv. Drug Delivery Rev., 2003, 55, 585–609 CrossRef CAS.
- J. Yang, A. R. Webb and G. A. Ameer, Novel Citric Acid-Based Biodegradable Elastomers for Tissue Engineering, Adv. Mater., 2004, 16(6), 511–516 CrossRef CAS.
- J. Yang, A. R. Webb, S. J. Pickerill, G. Hageman and G. A. Ameer, Synthesis and evaluation of poly(diol citrate) biodegradable elastomers, Biomaterials, 2006, 27(9), 1889–1898 CrossRef CAS.
- H. Zhao, M. C. Serrano, D. A. Popowich, M. R. Kibbe and G. A. Ameer, Biodegradable nitric oxide-releasing poly(diol citrate) elastomers, J. Biomed. Mater. Res., Part A, 2010, 93A(1), 356–363 CAS.
- R. Riva, P. Lussis, S. Lenoir, C. Jerome, R. Jerome and P. Lecomte, Contribution of "Click Chemistry" to the Synthesis of Antimicrobial Aliphatic Copolyester, Polymer, 2008, 49, 2023–2028 CrossRef CAS.
- P. N. Coneski, K. S. Rao and M. H. Schoenfisch, Degradable Nitric Oxide-Releasing Biomaterials via Post-Polymerization Functionalization of Cross-Linked Polyesters, Biomacromolecules, 2010, 11(11), 3208–3215 CrossRef CAS.
- C. K. Williams, Synthesis of Functionalized Biodegradable Polyesters, Chem. Soc. Rev., 2007, 36, 1573–1580 RSC.
- C. Jerome and P. Lecomte, Recent Advances in the Synthesis of Aliphatic Polyesters by Ring-Opening Polymerization, Adv. Drug Delivery Rev., 2008, 60, 1056–1076 CrossRef CAS.
- R. J. Pounder and A. P. Dove, Towards Poly(ester) Nanoparticles: Recent Advances in the Synthesis of Functional Poly(ester)s by Ring-Opening Polymerization, Polym. Chem., 2010, 1, 260–271 RSC.
- J. H. Wynne, P. A. Fulmer, D. M. McCluskey, N. M. Mackey and J. P. Buchanan, Synthesis and Development of a Multifunctional Self-Decontaminating Polyurethane Coating, ACS Appl. Mater. Interfaces, 2011, 3(6), 2005–2011 CAS.
- P. Kurt and K. J. Wynne, Co-Polyoxetanes with Alkylammonium and Fluorous or PEG-Like Side Chains:Soft Blocks for Surface Modifying Polyurethanes, Macromolecules, 2007, 40(26), 9537–9543 CrossRef CAS.
- S. B. Lee, R. R. Koepsel, S. W. Morley, K. Matyjaszewski, Y. Sun and A. J. Russell, Permanent, Nonleaching Antibacterial Surfaces. 1. Synthesis by Atom Transfer Radical Polymerization, Biomacromolecules, 2004, 5(3), 877–882 CrossRef CAS.
- P. N. Coneski, P. A. Fulmer and J. H. Wynne, Enhancing the Fouling Resistance of Biocidal Urethane Coatings via Surface Chemistry Modulation, Langmuir, 2012, 28(17), 7039–7048 CrossRef CAS.
- D. G. Barrett and M. N. Yousaf, Poly(triol α-ketoglutarate) as Biodegradable, Chemoselective, and Mechanically Tunable Elastomers, Macromolecules, 2008, 41(17), 6347–6352 CrossRef CAS.
- D. G. Barrett, W. Luo and M. N. Yousaf, Aliphatic polyester elastomers derived from erythritol and [small alpha],[small omega]-diacids, Polym. Chem., 2010, 1(3), 296–302 RSC.
- W. Xie, Z. Gao, W.-P. Pan, D. Hunter, A. Singh and R. Vaia, Thermal Degradation Chemistry of Alkyl Quaternary Ammonium Montmorillonite, Chem. Mater., 2001, 13(9), 2979–2990 CrossRef CAS.
- W. Yawei, S. Wenjian, Y. Yun, C. Xiaoyun, in Synthesization Characterization and Properties of Quaternary Ammonium Cationic Cellulose, Bioinformatics and Biomedical Engineering, ICBBE 2009, 3rd International Conference, 11–13 June 2009, 2009, pp. 1–4 Search PubMed.
- C. D. Bain and G. M. Whitesides, A study by contact angle of the acid–base behavior of monolayers containing .omega.-mercaptocarboxylic acids adsorbed on gold: an example of reactive spreading, Langmuir, 1989, 5(6), 1370–1378 CrossRef CAS.
- C. V. Benedict, W. J. Cook, P. Jarrett, J. A. Cameron, S. J. Huang and J. P. Bell, Fungal degradation of polycaprolactones, J. Appl. Polym. Sci., 1983, 28(1), 327–334 CrossRef CAS.
- H. Pranamuda, Y. Tokiwa and H. Tanaka, Microbial degradation of an aliphatic polyester with a high melting point, poly(tetramethylene succinate), Appl. Environ. Microbiol., 1995, 61(5), 1828–32 CAS.
- T. Suyama, Y. Tokiwa, P. Ouichanpagdee, T. Kanagawa and Y. Kamagata, Phylogenetic Affiliation of Soil Bacteria That Degrade Aliphatic Polyesters Available Commercially as Biodegradable Plastics, Appl. Environ. Microbiol., 1998, 64(12), 5008–5011 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Complete table of synthesized compositions, NMR spectra of QAS diols, thermal analysis data of additional poly(diol citrate) compositions, and kinetic plots of poly(diol citrate) hydrolysis. See DOI: 10.1039/c2ra21286b |
| This journal is © The Royal Society of Chemistry 2012 |