Nanoporous core–shell Cu@Cu2O nanocomposites with superior photocatalytic properties towards the degradation of methyl orange
Tianyi
Kou
a,
Chuanhong
Jin
b,
Chi
Zhang
a,
Junzhe
Sun
a and
Zhonghua
Zhang
*a
aKey Laboratory for Liquid–Solid Structural Evolution and Processing of Materials (Ministry of Education), School of Materials Science and Engineering, Center for Advanced Energy Materials and Technology Research, Shandong University, Jingshi Road 17923, Jinan, 250061, P. R. China. E-mail: zh_zhang@sdu.edu.cn
bCenter for Electron Microscopy, National Key laboratory for Silicon Materials, Department of Materials Science and Engineering, Zhejiang University, Hangzhou, 310027, P. R. China
First published on 16th October 2012
Abstract
We present a facile two-step strategy to fabricate a novel nanoporous core–shell Cu@Cu2O nanocomposite photocatalyst. The photocatalyst was synthesized via the combination of dealloying of the as-milled Al66.7Cu33.3 (at.%) precursor powders in alkaline media with subsequent surface oxidation in air. The microstructure of the as-prepared photocatalyst has been characterized by X-ray diffraction (XRD), a field-emission scanning electron microscope (FESEM) and a transmission electron microscope (TEM). The results show that the as-prepared photocatalyst exhibits an open, bicontinuous interpenetrating ligament (ca. 30 nm)–channel (ca. 15 nm) structure, and is comprised of Cu (core) and Cu2O (shell) phases. Methyl orange (MO) was used as the model pollutant, and photodegradation experiments were monitored by UV-vis spectrophotometry. The nanoporous core–shell Cu@Cu2O photocatalyst shows excellent photocatalytic activity towards the degradation of MO under sunlight. The MO degradation rate can reach as high as 8.04 mg min−1 gcat−1. In addition, the influence of additive amounts of photocatalyst and initial concentrations of MO on the photodegradation process has been investigated. The photodegradation mechanism of the as-prepared photocatalyst has also been discussed.
1. Introduction
Colorants are necessary chemicals in the textile industry,1 but most of them are detrimental to the environment and aquatic creatures if directly poured into water. It has been reported that more than 700![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons of dye-stuff are produced annually and about 20% of them are discharged by textile factories.2,3 Among these dyes and pigments, azo colorants are hard to degrade due to their complex chemical structures and synthetic origin.4 The traditional ways of processing wastewater containing azo colorants through coagulation-flocculation5 or activated carbon adsorption6 are inefficient since they just transfer the dye molecules from liquid to solid.7 In the past few years, photocatalysis has attracted much attention as a new and efficient method to treat wastewater containing colorants. Semiconductor-based photocatalysts have been widely studied, such as titanium dioxide (TiO2) and zinc oxide (ZnO).8,9 Unfortunately, TiO2 and ZnO can only absorb a small fraction of sunlight (ultraviolet or near ultraviolet) due to their broad band gap (3.0–3.2 eV).8 Therefore, it is urgent to develop photocatalysts which can effectively utilize solar energy.
000 tons of dye-stuff are produced annually and about 20% of them are discharged by textile factories.2,3 Among these dyes and pigments, azo colorants are hard to degrade due to their complex chemical structures and synthetic origin.4 The traditional ways of processing wastewater containing azo colorants through coagulation-flocculation5 or activated carbon adsorption6 are inefficient since they just transfer the dye molecules from liquid to solid.7 In the past few years, photocatalysis has attracted much attention as a new and efficient method to treat wastewater containing colorants. Semiconductor-based photocatalysts have been widely studied, such as titanium dioxide (TiO2) and zinc oxide (ZnO).8,9 Unfortunately, TiO2 and ZnO can only absorb a small fraction of sunlight (ultraviolet or near ultraviolet) due to their broad band gap (3.0–3.2 eV).8 Therefore, it is urgent to develop photocatalysts which can effectively utilize solar energy.
Cuprous oxide (Cu2O) is a p-type semiconductor with a direct band gap of 2.0–2.2 eV.10 Hence, Cu2O can absorb a large part of visible light (λ < 620 nm) and is a promising photocatalyst to degrade colorants. Cu2O has been prepared through electrolysis, the reduction of copper (Cu) II salts, and a hydrothermal method.11–13 In the past few years, various Cu2O photocatalysts with micro-/nano-structures have been synthesized, such as nanocubes,14 octahedra,15 multipods,16etc. However, photoelectrons easily tend to recombine with holes in Cu2O-based photocatalysts, which leads to the limitation of their photocatalytic activities.17 According to the previous studies, the heterojunction of Cu and Cu2O can enhance the photocatalytic property of Cu2O-based semiconductors, because the existence of Cu can promptly transfer photoelectrons, avoiding the recombination of electron–hole pairs.18
Recently, nanoporous metals have attracted more and more interest due to their high specific surface area and unique bicontinuous ligament–channel structures, and show promising applications in fields of catalysis,19 sensors,20 actuators,21 fuel cells,22 and so on. For example, Xu et al.23 have reported that nanoporous gold (NPG) with ligament sizes less than 6 nm show excellent catalytic activities towards CO oxidation at low temperatures. Dealloying is a common and effective method to obtain nanoporous metals, originally used in depletion gilding treatment to colorate artifacts during the Inca civilization period.24 It refers to the selective dissolution of one or more active elements out of the precursor alloys and can be realized through electrochemical or chemical corrosion, leading to a biocontinuous interpenetrating ligament–channel structure.25 In addition, nanoporous metals are good substrate materials to construct bimetallic or metal/polymer nanocomposites due to their monolithic morphology and excellent electrical conductivity. Through a simple immersion–electrodeposition (IE) method, a tiny amount of Pt can be deposited in a quasi-two-dimensional form onto the NPG substrate, forming nanostructured bimetallic Pt–Au catalysts.26 Meng and Ding27 recently reported the design and facile fabrication of an ultrathin flexible all-solid-state supercapacitor based on a polypyrrole (PPy)-decorated NPG leaf nanocomposite through a dealloying and electropolymerization process. Compared to Au, Cu is much cheaper and nanoporous Cu has been fabricated through dealloying Mn–Cu and Al–Cu precursors.28,29 To the best of our knowledge, however, no information is available on the photocatalytic applications of nanoporous Cu.
In this work, we aimed at the design and fabrication of a novel nanoporous core–shell Cu@Cu2O photocatalyst through a facile two-step strategy involving dealloying and surface oxidation. Nanoporous Cu powders were firstly prepared by dealloying the Al–Cu precursor in alkaline media. Subsequently, surface oxidation resulted in the formation of nanoporous core–shell Cu@Cu2O nanocomposites. In photodegradation experiments, methyl orange (MO) was used as the model pollutant because it is a water-soluble azo colorant and is widely used in textile, printing, paper manufacturing, pharmaceutical and food industries.30 The effect of additive amounts of photocatalyst and MO concentrations on the photocatalytic activity has been investigated at ambient temperature. The photodegradation results have shown that our Cu@Cu2O photocatalyst exhibits an excellent photocatalytic activity.
2. Experimental section
The Al66.7Cu33.3 precursor alloy was prepared from elemental powders by ball milling in a Fritsch P6 mill. The dealloying of the as-milled Al–Cu powders (0.5 g) was performed in a 100 mL beaker containing 2 mol L−1 NaOH aqueous solution for 1.5 h at ambient temperature. Then the as-dealloyed samples were rinsed by ultra-pure water and dehydrated alcohol (analytical grade). The as-dealloyed samples were kept in a vacuum drying tank for 10 h, and the photocatalyst was obtained by the following surface oxidation in air at ambient temperature for 2 h. The phase constitution of the as-prepared photocatalyst was characterized by X-ray diffraction (XRD, Rigaku D/max-rB) with Cu-Kα radiation. The field-emission scanning electron microscope (FESEM, Quanta FEG 250) and transmission electron microscope (TEM, Hitachi H 9500) were employed to study the morphology and nanoporous structure of the as-prepared photocatalyst.To study the effect of different additive amounts of photocatalyst and the initial concentration of MO on the photocatalytic activity of the as-prepared photocatalyst, we designed four systems as follows: (I) photocatalyst (4 mg) + MO (20 mg L−1), (II) photocatalyst (6 mg) + MO (20 mg L−1), (III) photocatalyst (8 mg) + MO (20 mg L−1), and (IV) photocatalyst (8 mg) + MO (40 mg L−1). To eliminate the effect of solar radiation at different times on our photocatalytic results, the degradation experiments were carried out in a fixed time range during the day (11:30 to 14:30). Taking system (IV) for example, the as-prepared photocatalyst (8 mg) was dispersed in a 100 mL beaker containing 50 mL MO aqueous solution (40 mg L−1), and was stirred in the dark for 20 min to achieve adsorption saturation. Then the suspension was exposed to sunlight and the photodegradation started. The photodegradation experiments were carried out under magnetic stirring conditions at ambient temperature. A small amount of MO solution (5 mL) was extracted at certain intervals and the concentration of MO was monitored by an UV-vis spectrophotometer (Mapada) at 465 nm, which is the maximum absorbance wavelength of MO. The photodegradation ratio (D) was calculated according to the following equation: D = (A0 − A)/A0 × 100%, where A0 is the original absorbance of MO at its maximum absorbance wavelength and A is the absorbance of MO at the same wavelength after different degradation durations.
3. Results and discussion
3.1 Fabrication and characterization of photocatalyst
Fig. 1 shows the XRD pattern of the as-prepared photocatalyst. There are three major diffraction peaks (2θ = 43.3, 50.4 and 74.1°) in the XRD pattern, corresponding to the (111), (200) and (220) reflections of a face centered cubic (f.c.c.) Cu (PDF # 04–0836) respectively. In addition, there are three minor diffraction peaks (2θ = 29.6, 36.5 and 61.5°) which can be indexed to Cu2O (PDF # 65–3288), corresponding to the (110), (111) and (220) reflections respectively. It should be noted that other diffraction peaks of Cu2O at 42.4 and 72.7° overlap with the (111) and (220) reflections of Cu. Therefore, the as-obtained photocatalyst is composed of Cu and Cu2O phases, and Cu is dominant according to the relative intensities of both phases (Fig. 1).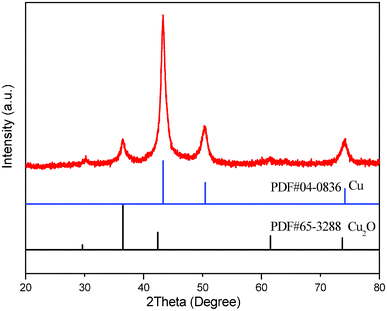 | ||
| Fig. 1 XRD pattern of the as-prepared photocatalyst obtained through dealloying of the as-milled Al66.7Cu33.3 powders in the 2 mol L−1 NaOH aqueous solution and subsequent surface oxidation in air. | ||
Fig. 2 shows the particle size distribution, morphology and nanoporous structure of the as-prepared photocatalyst. The size of the as-prepared photocatalyst particles ranges from several hundred nanometers (ca. 300 nm) to several micrometers (ca. 4 μm) (Fig. 2a). Fig. 2b shows the morphology of one particle and the nanoporous structure can be observed at a higher magnification. As shown in Fig. 2c, the bicontinuous interpenetrating ligament–channel structure can be clearly seen. The size of channels is ca. 15 nm, and that of ligaments is ca. 30 nm. The energy dispersive X-ray (EDX) results demonstrate that the as-prepared photocatalyst is mainly composed of Cu, O and a minor amount of residual Al. A typical EDX spectrum is presented in Fig. 2d.
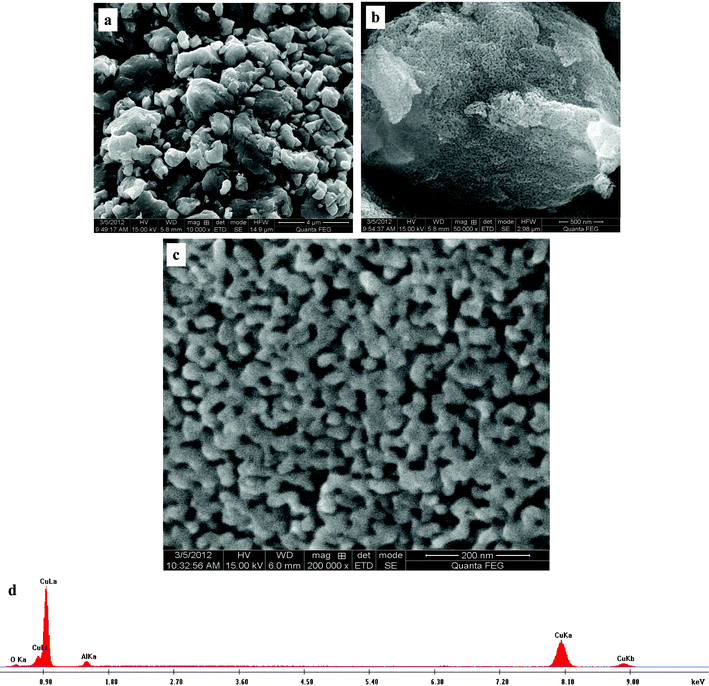 | ||
| Fig. 2 FESEM images showing (a) the particle size distribution, (b) the morphology of the particles and (c) the nanoporous structure of the as-prepared photocatalyst. (d) A typical EDX spectrum. | ||
The TEM results further confirm the nanoporous structure of the as-prepared photocatalyst (Fig. 3). The typical TEM bright-field image (Fig. 3a) clearly shows that the photocatalyst exhibits an open bicontinuous ligament–channel structure, which is consistent with the FESEM observation. Fig. 3b shows the corresponding selected-area electron diffraction (SAED) pattern of the nanoporous area of the as-prepared photocatalyst. The SAED pattern is composed of polycrystalline diffraction rings, indicating the nanocrystalline nature of the ligaments in the as-prepared photocatalyst. The strong rings correspond well to the (111), (200), (220) and (311) reflections of f.c.c. Cu, as marked by the solid arrows in Fig. 3b. Besides, the weak rings can be indexed as the (111) and (220) reflections of Cu2O, as highlighted by dotted arrows in Fig. 3b. The electron diffraction results further verify that the as-prepared photocatalyst is comprised of Cu and Cu2O. Fig. 3c shows one typical high-resolution TEM (HRTEM) image, and some lattice fringes can be clearly seen. However, it is difficult to distinguish Cu from Cu2O through the observation of the lattice fringes. The interplanar spacing of 2.08 Å corresponds to the (111) reflection of f.c.c. Cu, as marked in Fig. 3c. It should be noted that the spacing of 2.08 Å is also close to the 2.13 Å of the (200) reflection of Cu2O. In spite of this, the surface layer should be Cu2O covering the Cu ligaments.
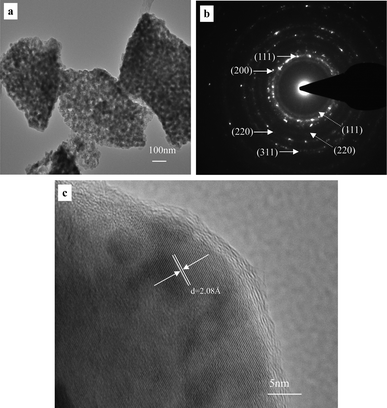 | ||
| Fig. 3 (a) TEM image showing the nanoporous structure of the as-prepared photocatalyst, (b) the corresponding SAED pattern, and (c) a typical HRTEM image showing lattice fringes of one ligament. | ||
As previously described, the nanoporous core–shell Cu@Cu2O nanocomposites can be obtained through the combination of dealloying with surface oxidation. The formation mechanism of these nanocomposites can be depicted as follows, and is schematically shown in Fig. 4a. It is generally recognized that the dealloying process involves the dissolution of the less noble element (here, Al) and the formation/coarsening of the nanoporous ligament–channel structure by surface diffusion of the more noble element (here, Cu).24,25 During dealloying, Al is preferentially dissolved into the electrolyte through the formation of AlO2−. Thus the released Cu atoms become adatoms, which diffuse along the alloy/electrolyte interface and re-organize into the ligament–channel structure, as shown in the left part of Fig. 4a. Subsequently, the surface of the Cu ligaments (core) is oxidized by the O2 in air to form the Cu2O layer (shell), as illustrated in the right part of Fig. 4a. Due to the similar interplanar spacing of Cu (111) and Cu2O (200), it is reasonable to assume that the Cu2O surface layer can form through epitaxial growth on the Cu lattice. Finally, the nanoporous core–shell structure forms through the two-step dealloying–oxidation process. Furthermore, we can modulate the thickness (or amount) of the Cu2O shell in the Cu@Cu2O nanocomposites through control over the oxidation duration during the second step.
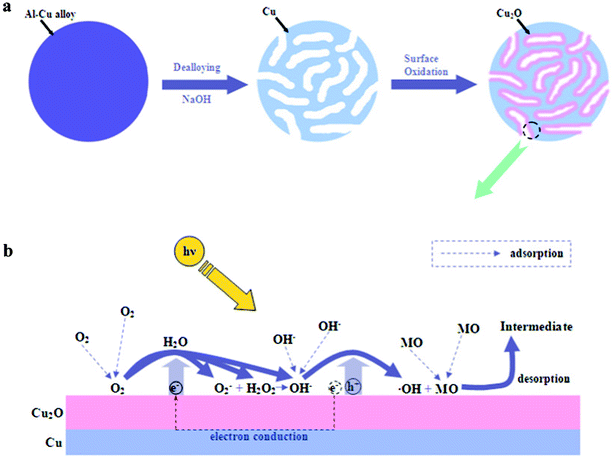 | ||
| Fig. 4 The schematic illustration showing (a) the formation mechanism of the nanoporous core–shell Cu@Cu2O photocatalyst and (b) the photodegradation mechanism of MO. | ||
3.2 Assessment of photocatalytic activity
The photocatalytic performance of the as-prepared photocatalyst was evaluated by photodegradation of MO under sunlight irradiation. We firstly discuss the influence of additive amounts of photocatalyst on the degradation process. Fig. 5 shows the relationship between the photodegradation ratio and the irradiation time of system I–III, and the corresponding UV-vis absorption spectra are presented as insets. As shown in the inset of Fig. 5a, the UV-vis absorption spectra demonstrate that the intensity of the characteristic absorbance peak of MO at its maximum absorbance wavelength (λ = 465 nm) decreases with increasing irradiation time in system I (4 mg photocatalyst in the 20 mg L−1 MO solution). After 100 min, about 90% of MO was degraded, which can be seen from the corresponding plot of photodegradation ratio versus irradiation time (Fig. 5a). Similar scenarios can be found in the photodegradation process of the other two systems (system II and III: 6 and 8 mg photocatalyst in the 20 mg L−1 MO solution), as shown in Fig. 5b and 5c respectively. However, the degradation rate is quite different for different additive amounts of photocatalyst. A duration of 35 min is necessary for system II to achieve a photodegradation ratio of 90% (Fig. 5b), but only 14 min is needed for system III (Fig. 5c). Therefore, the degradation rate greatly accelerates and the duration needed to finish the degradation process sharply shortens with increasing the additive amount of photocatalyst.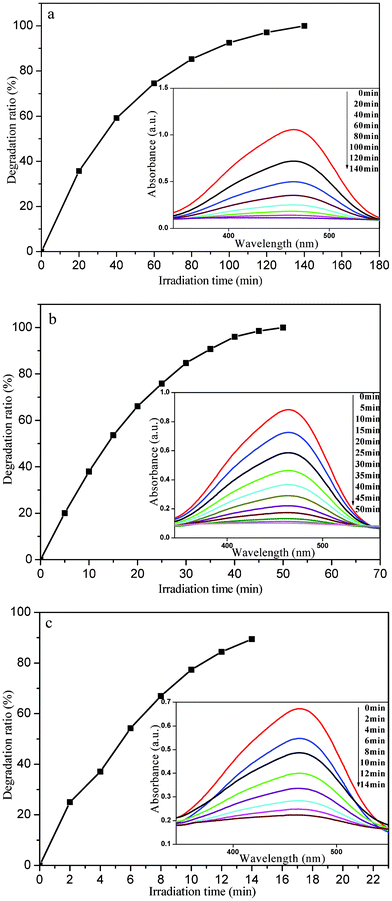 | ||
| Fig. 5 The plots of photodegradation ratio versus irradiation time of system I–III for photocatalytic degradation of MO by the nanoporous core–shell Cu@Cu2O photocatalyst (a: system I; b: system II; c: system III). Insets: corresponding UV-vis absorption spectra. | ||
In addition, we also investigated the effect of different initial concentrations of MO on the photocatalytic activity of the present photocatalyst. Fig. 6 shows the relationship between the photodegradation ratio and irradiation time of system IV (8 mg photocatalyst + 40 mg L−1 MO solution), and the corresponding UV-vis absorption spectra are presented in the inset. It is clear that the degradation process and the evolution of the absorption spectra are similar to those for system I–III (Fig. 5). The duration required to finish the degradation process is about 140 min, much longer than that for system III (8 mg photocatalyst + 20 mg L−1 MO solution, Fig. 5c). According to the absorption spectra, 110 min has been used when 90% of MO was degraded, which is almost 8 times that for the 8 mg photocatalyst to achieve a photodegradation ratio of 90% in the 20 mg L−1 MO solution.
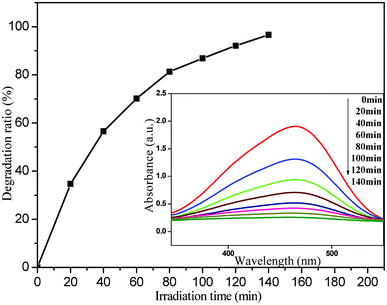 | ||
| Fig. 6 The plot of photodegradation ratio versus irradiation time of system IV (8 mg photocatalyst + 40 mg L−1 MO) for photocatalytic degradation of MO by the nanoporous core–shell Cu@Cu2O photocatalyst. Inset: corresponding UV-vis absorption spectra. | ||
The phenomena mentioned above can be explained as follows, and the mechanism of photodegradation is depicted in Fig. 4b. When exposed to the sunlight, the Cu2O semiconductor oxide can be excited to produce electrons and holes. The photo-generated electrons and holes can initiate a series of photodegradation reactions. Holes at the valence band can oxidize hydroxyl ions (OH−) adsorbed on the surface of the photocatalyst to yield hydroxyl radicals (·OH) which play an important role in photodegradation.31 ·OH has a higher redox potential (+ 1.9 V)32 than that of MO (+ 0.94 V),33 which is why ·OH rather than the photo-generated holes (+ 0.519 V) can oxidize adsorbed MO.34 On the other hand, the photo-generated electrons conducted away from holes by Cu can be captured by adsorbed oxygen molecules (O2), leading to the generation of hydrogen peroxide (H2O2) and OH−, or super oxide radical ions (O2−).35 O2− can further interact with H2O2, which facilitates the yield of OH− and ·OH and in return contributes to the photodegradation process of MO.34 Finally, MO can be oxidized into intermediates and be desorbed from the surface of Cu2O.
By means of increasing the additive amount of the photocatalyst, the photocatalytic activity and degradation rate can be greatly enhanced at the given initial concentration of MO (Fig. 5). This can be explained as follows: the number of active sites for producing ·OH on the surface of the photocatalyst increases with increasing additive amount, which can facilitate the process of photodegradation. Therefore, the time to reach the same photodegradation ratio sharply decreases with an increasing additive amount of the photocatalyst from 4 to 8 mg in the 20 mg L−1 MO solution. When the initial concentration of MO increases from 20 to 40 mg L−1, the irradiation time to achieve 90% of the photodegradation ratio also increases from 14 to 110 min. When increasing the initial concentration of MO, the active sites that can adsorb OH− will be largely occupied by MO molecules, which leads to the decrease in producing ·OH and thereby exerts a negative effect on the photodegradation process.36 Consequently, by varying the initial concentration of MO from 20 to 40 mg L−1, the retardation degree in the penetration of light will be enhanced.36 Thus the utilization ratio of sunlight energy will decrease in the photodegradation process, resulting in the decrease of the photocatalytic activity of the as-prepared photocatalyst. However, it should be noted that the photodegradation rate would increase with increasing initial MO concentration in the low concentration region because the light interception by MO is negligible in the case.
The MO degradation rate can roughly reflect the degradation efficiency of the as-prepared photocatalyst. The estimated values for the MO degradation rates in systems I–III range from ∼2.25–∼8.04 (mg min−1 gcat−1), which are greatly larger than those of the recently reported photocatalysts under similar degradation conditions (Table 1).18,37,38 The experimental data of photodegradation for systems I–III can be well fitted by a pseudo-first-order kinetic model (Fig. 7), in which the kinetic equation was used to describe the process:
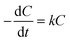 | (1) |
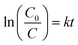 | (2) |
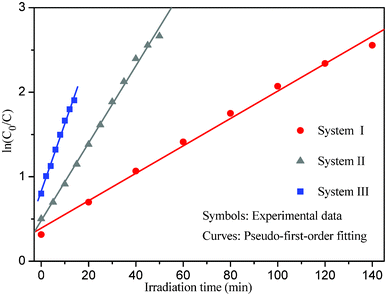 | ||
| Fig. 7 Plots of the pseudo-first-order kinetic model for systems I–III. | ||
| Photocatalyst | Additive amount (mg) | MO concentration (mg L−1) | Degradation ratio (%) | Irradiation time (min) | Degradation rate (mg min−1 gcat−1) | Light source |
|---|---|---|---|---|---|---|
| Core–shell Cu@Cu2O (this work) | 4 | 20 | 90 | 100 | 2.25 | Sun light (300–2500 nm) |
| 6 | 35 | 4.29 | ||||
| 8 | 14 | 8.04 | ||||
| Porous AgCl@Ag nanocomposites37 | 10 | 10 | 90 | 60 | 0.75 | 300 W Xe arc lamp (λ > 420 nm) |
| Cu2O nanocube38 | 8 | 20 | 83.6 | 120 | 0.70 | 300 W xenon lamp (190–1100 nm) |
| Cu2O@Cu18 | 30 | 10 | 90 | 120 | 0.14 | 40 W tungsten lamp (350–2500 nm) |
Adsorption is an important precondition for the whole photodegradation process, while diffusion of the involved species (MO, OH−, intermediate, O2, etc.) plays a key role in the adsorption stage. Fig. 8 shows the relationship between the photodegradation ratio and the square root of the irradiation time for systems I–IV during the photocatalytic degradation of MO by the nanoporous core–shell Cu@Cu2O photocatalyst. It is obvious that good linear correlations can be obtained for the photodegradation process of systems I–IV. Thus it is reasonable to assume that the photodegradation of MO by the present photocatalyst is a diffusion-controlled process.39,40 In addition, the slope of the fitted lines can also reflect the photodegradation rate or efficiency. From Fig. 8a–c, the slopes of the fitted lines were determined to be 1.23209, 2.45174, and 3.79107 for system I–III respectively. Obviously, the slope increases with the increasing additive amount of the as-prepared photocatalyst, which means that the higher slope value corresponds to a faster photodegradation process. As shown in Fig. 8d, system IV has a slope of 1.07396, which is much lower than that of system III. This means that the photodegradation rate markedly decreases with increasing initial concentration of MO for the present concentration region.
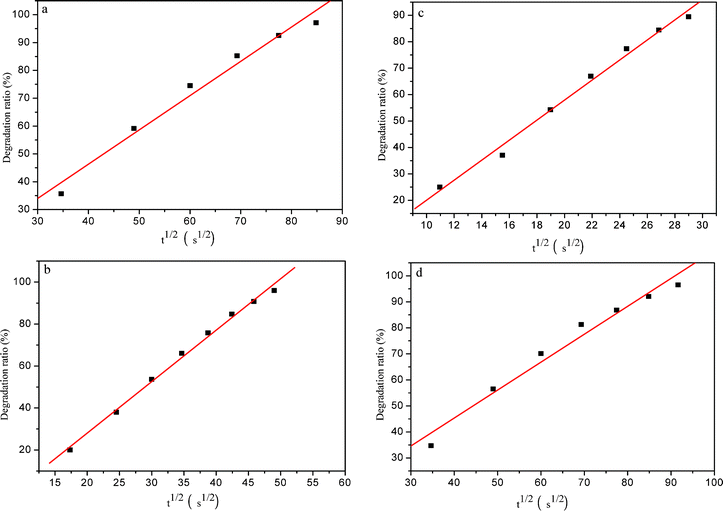 | ||
| Fig. 8 The plots of photodegradation ratio versus the square root of irradiation time for system I–IV (a–d) for the photocatalytic degradation of MO by the nanoporous core–shell Cu@Cu2O photocatalyst. | ||
4. Conclusions
In summary, a novel nanoporous core–shell Cu@Cu2O nanocomposite photocatalyst can be synthesized through a facile two-step strategy: dealloying of the as-milled Al66.7Cu33.3 (at.%) precursor powders in the NaOH aqueous solution and subsequent surface oxidation in air. This Cu@Cu2O photocatalyst shows superior photocatalytic activity towards the degradation of MO under sunlight, which is due to its unique nanoporous core–shell nanocomposite structure. Moreover, both the additive amounts of the photocatalyst and the initial concentration of MO have a significant effect on the photodegradation process and the photocatalytic activity. The photodegradation rate markedly increases with an increase in additive amount of photocatalyst or a decrease in the initial concentration of MO. In addition, the good linear correlation between the photodegradation ratio and the square root of the irradiation time demonstrates that the photodegradation of MO by the present photocatalyst is a diffusion-controlled process. Based upon our findings, novel nanoporous core–shell photocatalysts can be designed and fabricated through the dealloying route in combination with other methods like surface oxidation.Acknowledgements
The authors gratefully acknowledge financial support by Program for New Century Excellent Talents in University (MOE), National Basic Research Program of China (973, 2012CB932800), Independent Innovation Foundation of Shandong University (2010JQ015) and National Natural Science Foundation of China (50971079).References
- K. Golka, S. Kopps and Z. W. Myslak, Toxicol. Lett., 2004, 151, 203 CrossRef CAS.
- T. Robinson, G. McMullan, R. Marchant and P. Nigam, Bioresour. Technol., 2001, 77, 247 CrossRef CAS.
- I. Arslan and I. A. Balcioglu, Chemosphere, 1999, 39, 2767 CrossRef CAS.
- P. A. Carneiro, R. F. P. Nogueira and M. V. B. Zanoni, Dyes Pigm., 2007, 74, 127 CrossRef CAS.
- L. Semerjian and G. M. Ayoub, Adv. Environ. Res., 2003, 7, 389 CrossRef CAS.
- A. Wang, J. Qu, J. Ru, H. Liu and J. Ge, Dyes Pigm., 2005, 65, 227 CrossRef CAS.
- M. A. Behnajady, N. Modirshahla, N. Daneshvar and M. Rabbani, Chem. Eng. J., 2007, 127, 167 CrossRef CAS.
- D. Jiang, Y. Xu, D. Wu and Y. Sun, J. Solid State Chem., 2008, 181, 593 CrossRef CAS.
- A. O. Musa, T. Akomolafe and M. J. Carter, Sol. Energy Mater. Sol. Cells, 1998, 51, 305 CrossRef CAS.
- I. Grozdanov, Mater. Lett., 1994, 19, 281 CrossRef CAS.
- H. Yang, J. Ouyang, A. Tang, Y. Xiao, X. Li, X. Dong and Y. Yu, Mater. Res. Bull., 2006, 41, 1310 CrossRef CAS.
- X. Zhang, J. Song, J. Jiao and X. Mei, Solid State Sci., 2010, 12, 1215 CrossRef CAS.
- Z. Chen, E. Shi, Y. Zheng, W. Li, B. Xiao and J. Zhuang, J. Cryst. Growth, 2003, 249, 294 CrossRef CAS.
- L. Gou and C. J. Murphy, Nano Lett., 2003, 3, 231 CrossRef CAS.
- M. J. Siegfried and K. S. Choi, J. Am. Chem. Soc., 2006, 128, 10356 CrossRef CAS.
- H. Pang, F. Gao and Q. Lu, Chem. Commun., 2009, 1076 RSC.
- Y. Tang, Z. Chen, Z. Jia, L. Zhang and J. Li, Mater. Lett., 2005, 59, 434 CrossRef CAS.
- B. Zhou, Z. Liu, H. Wang, Y. Yang and W. Su, Catal. Lett., 2009, 132, 75 CrossRef CAS.
- G. C. Bond and D. T. Thompson, Catal. Rev., 1999, 41, 319 CAS.
- T. You, O. Niwa, M. Tomita and S. Hirono, Anal. Chem., 2003, 75, 2080 CrossRef CAS.
- J. Weissmueller, R. N. Viswanath, D. Kramer, P. Zimmer, R. Wuerschum and H. Gleiter, Science, 2003, 300, 312 CrossRef CAS.
- S. H. Joo, S. J. Choi, K. J. Kwa and Z. Liu, Nature, 2001, 412, 169 CrossRef CAS.
- C. Xu, J. Su, X. Xu, P. Liu, H. Zhao, F. Tian and Y. Ding, J. Am. Chem. Soc., 2007, 129, 42 CrossRef CAS.
- A. J. Forty, Nature, 1979, 282, 597 CrossRef CAS.
- J. Erlebacher, M. J. Aziz, A. Karma, N. Dimitrov and K. Sieradzki, Nature, 2001, 410, 450 CrossRef CAS.
- J. Zhang, H. Ma, D. Zhang, P. Liu, F. Tian and Y. Ding, Phys. Chem. Chem. Phys., 2008, 10, 3250 RSC.
- F. Meng and Y. Ding, Adv. Mater., 2011, 23, 4098 CrossRef CAS.
- J. R. Hayes, A. M. Hodge, J. Biener, A. V. Hamza and K. Sieradzki, J. Mater. Res., 2006, 21, 2611 CrossRef CAS.
- Z. Qi, C. Zhao, X. Wang, J. Lin, W. Shao, Z. Zhang and X. Bian, J. Phys. Chem. C, 2009, 113, 6694 CAS.
- S. C. Rastogi, V. J. Barwick and S. V. Carter, Chromatographia, 1997, 45, 215 CAS.
- Z. Zheng, B. Huang, Z. Wang, M. Guo, X. Qin, X. Zhang, P. Wang and Y. Dai, J. Phys. Chem. C, 2009, 113, 14448 CAS.
- V. Subramanian, E. Wolf and P. V. Kamat, J. Phys. Chem. B, 2001, 105, 11439 CrossRef CAS.
- M. Louis and Z. Petr, CRC Handbook Series in Organic Electrochemistry, CRC Press, Ohio, 1977, vol. 1 Search PubMed.
- L. Huang, F. Peng, H. Yu and H. Wang, Solid State Sci., 2009, 11, 129 CrossRef CAS.
- A. P. L. Batista, H. W. P. Carvalho, G. H. P. Luz, P. F. Q. Martins, M. Goncalves and L. C. A. Oliveira, Environ. Chem. Lett., 2010, 8, 63 CrossRef CAS.
- A. Tang, Y. Xiao, J. Ouyang and S. Nie, J. Alloys Compd., 2008, 457, 447 CrossRef CAS.
- Y. Li and Y. Ding, J. Phys. Chem. C, 2010, 114, 3175 CAS.
- W. Sun, W. Sun, Y. Zhuo and Y. Chu, J. Solid State Chem., 2011, 184, 1638 CrossRef CAS.
- W. J. Weber and J. C. Morris, Advances in water pollution research: removal of bio-logically resistant pollutant from waste water by adsorption, in: Proceedings of 1st International Conference on Water Pollution Symposium, Pergamon Press, Oxford, 1962, vol. 2, pp. 231 Search PubMed.
- W. J. Weber and J. C. Morris, Kinetics of adsorption on carbon from solutions, J. Sanit. Eng. Div. Am. Soc. Civ. Eng., 1963, 89, 31 Search PubMed.
| This journal is © The Royal Society of Chemistry 2012 |