Synthesis, unambiguous chemical characterization, and reactivity of 2,3,4,5-tetrahydro-1,5-benzoxazepines-3-ol†
M. Eugenia
García-Rubiño
,
María C.
Núñez
,
Miguel A.
Gallo
and
Joaquín M.
Campos
*
Departamento de Química Farmacéutica y Orgánica, Facultad de Farmacia, c/Campus de Cartuja s/n, 18071 Granada, Spain. E-mail: jmcampos@ugr.es; Fax: +34 958 243845; Tel: +34 958 243850
First published on 22nd October 2012
Abstract
The previous structures of several 3,4-dihydro-2H-3-hydroxymethyl-1,4-benzoxazines were erroneous (Bioorg. Med. Chem. Lett. 2006, 16, 2862; Synlett 2009, 3283 and Chinese Patent 2006-10043245). The results presented here are both detailed NMR experiments and synthetic aspects on this subject. We redress some points for the preparation of intermediates of the target molecules.
Introduction
The 2,3-dihydro-1,4-benzodioxin ring 1a (Fig. 1) constitutes an important skeletal fragment in medicinal chemistry.1 We have recently reviewed the chemistry of 2,3-dihydro-1,4 benzodioxins and bioisosteres as structural motifs for biologically active compounds.2 We used the bioisosteres 3,4-dihydro-2H-1,5-benzoxathiepine-3-ol 2 and 2,3-dihydro-1,4-benzoxathiine-2-methanol 3 (Fig. 1) as intermediates to obtain a series of substituted-9-(2,3-dihydro-1,4-benzoxathiine-3-and -2-ylmethyl) purine derivatives with notable anti-proliferative activities.3,4 Benzoxazepines are a well-known pharmacophore in medicinal chemistry showing promising activity against various diseases such as antipsychotic, central nervous system activity along with anti-cancer profile against breast cancer cells.5,6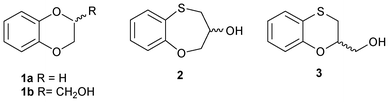 | ||
| Fig. 1 Structures of benzo-fused six- and seven-membered derivatives. | ||
Nitrogen-containing compounds are important chemical entities, many of which show unique bioactivity. 3,4-Dihydro-2H-1,4-benzoxazines have been used as a substructure of several biologically interesting agents between them.7–12
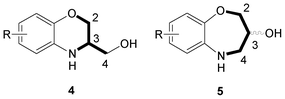
Following a bioisosteric replacement as a useful strategy for the structural modification of compounds 1b,132 and 3, we wish to report herein the synthesis and the correct chemical characterization of compounds 4a–f and 5a–c. We will discuss the misunderstanding in the scientific bibliography between the six-membered structures 4a–f and the seven-membered ones 5a–c, and the flaws encountered by us in the synthetic protocols that lead to both intermediates 4a and 5a and target molecules. Finally, we describe the Mitsunobu reaction3 of 5a with several substituted purines to obtain 4d–f (Fig. 2).
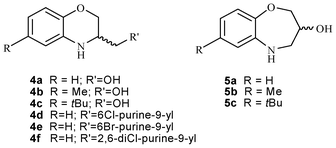 | ||
| Fig. 2 Molecules discussed in this manuscript. | ||
Results and discussion
After a search in literature we found two manuscripts dealing with the synthesis of 3,4-dihydro-2H-3-hydroxymethyl-1,4-benzoxazine (4a): (a) from 6 in the presence of Hantzsch 1,4-dihydropyridine (HEH) and Pd/C as catalyst14 (84%); (b) through a tandem reduction-oxirane opening of 1,2-epoxy-3-(o-nitrophenoxy)propane (6), in the presence of iron powder and acetic acid at reflux,15 or using Fe or Zn powder as reducing agent, and acetic acid or ammonium chloride as the acidic medium at the reflux temperature of H2O/EtOH.16 According to them,14–16 the nitro group was reduced, leading to the attack of the amino group on the epoxide moiety in a 6-exo-tet fashion, according to the Baldwin rules.17The preparation of 6 was published by Xu et al.18 Although they claim that 6 was obtained as a light yellow solid in a 35% yield after recrystallization (EtOH), we did not reproduce their results on repeating their experimental conditions and 7 (82%) was obtained instead as a yellow syrup (Scheme 1). Finally, 7 was subjected to an epoxide-forming process by using sodium hydride (1 equiv.) in anhydrous THF under an inert atmosphere (Scheme 1) and a very pure compound 6 (73%) was obtained as a light yellow solid. The synthetic procedure reported19 to 6 is different than that stated previously.
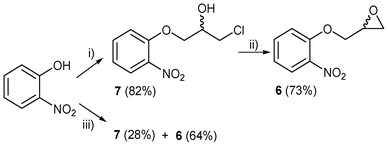 | ||
| Scheme 1 Reactivity of o-nitrophenol: (i) Conditions previously reported:18 epichlorohydrin (5 equiv.), NaOH (0.25 equiv.), deionized H2O, 85 °C, 2 h; (ii) NaH (1 equiv.), anhydrous THF, rt, instantaneous reaction; (iii) epichlorohydrin (5 equiv.), NaOH (1 equiv.), deionized H2O, 85 °C, 36 h. | ||
As we were surprised by the fact that Xu et al.18 only used 4 fold-less equivalents of the base in relation to o-nitrophenol, we decided to equalize both equivalents (1 equiv. of both o-nitrophenol and NaOH, but keep 5 equiv. of epichlorohydrin), and to increase the reaction time to 36 h, maintaining the reaction temperature at 85 °C (Scheme 1). The expected compound 6 was obtained in a fairly good yield (64%), together with a low yield (28%) of the propylene chlorohydrin 7.
In order to speed up the process of formation of 6, we used microwave irradiation and we obtained 7 (25%) and 6 (64%). When epichlorohydrin and o-nitrophenol were reacted in equimolecular quantities under MW irradiation at 95 °C for 40 min, the product of the reaction was the commercially available 1,3-bis-(o-nitrophenoxy)-2-propanol (8, 83%).
Nevertheless, the two following facts drew our attention after the cyclization reaction from epoxide 6: (a) two compounds were obtained, whilst the exclusive formation of w4a15 was published (See Scheme 2 to understand this terminology) in 84% yield; and (b) the 1H NMR spectrum of the mixture shows the presence of two isomers, as demonstrated by the presence of several sets of signals, each one composed of two splitting patterns.
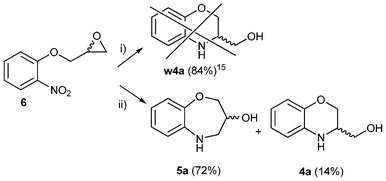 | ||
| Scheme 2 Regioselective reaction of the seven-membered ring 5a: i) according to reference 15 ii) Pd/C, EtOH, reflux, 12 h, inert atmosphere. The identification tag w4a15 denotes the wrong 4a with its bibliographic reference as superscript: it means “the incorrect structure 4a, described in the corresponding ref. 14, 15 or 16 but that is actually 5a”. | ||
The major isomer was identified as 2,3,4,5-tetrahydro-1,5-benzoxazepine-3-ol (5a, 72%) and the minor one was 3,4-dihydro-2H-3-hydroxymethyl-1,4-benzoxazine (4a, 14%) (the ratio between 5a/4a is 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). We then decided to carry out a shorter synthesis of 4a and 5a in a one-pot process. Starting from o-aminophenol (1 equiv.) and epichlorohydrin (1 equiv.), heating under MW-assisted conditions (160 °C, 30 min) produced a mixture of 4a (34%) and 5a (55%). Our complete 1H and 13C assignment is based on 1H signal integrals, multiplicity patterns, a DEPT(135) experiment, and HSQC/HMBC correlations. As the assignment is a routine task, only those points which are crucial to the unambiguous assignment of the previously incorrect structure are emphasized.
:1). We then decided to carry out a shorter synthesis of 4a and 5a in a one-pot process. Starting from o-aminophenol (1 equiv.) and epichlorohydrin (1 equiv.), heating under MW-assisted conditions (160 °C, 30 min) produced a mixture of 4a (34%) and 5a (55%). Our complete 1H and 13C assignment is based on 1H signal integrals, multiplicity patterns, a DEPT(135) experiment, and HSQC/HMBC correlations. As the assignment is a routine task, only those points which are crucial to the unambiguous assignment of the previously incorrect structure are emphasized.
The separation of 4a and 5a was carried out by HPLC [a mixture of H2O/MeCN (90/10) as eluent], semi-preparative column Nova-Pak C-18, 60 Å, 6 μm, 17.8 mm × 300 mm, retention time (tR) of 4a: 6.96 min, and tR of 5a: 7.27 min. The 1H and 13C NMR spectra (500 MHz, CDCl3), showed an important mistake, which is one of the key problems for the incorrect structure assignment of w4a.14–16 Zhao, Miao et al.15 literally state the following: The structure ofw4a14was confirmed by1H NMR and13C NMR data, which showed the presence of two CH2–O signals at δ 75.2 and δ 68.4 ppm, and a CH–N signal at δ 52.2 ppm (assigned to C-3), confirming the formation of only one regioisomer. The one-pot synthesis provides a novel and facile method of preparing the 2,3-dihydro-3-hydroxymethyl-1,4-benzoxazine derivatives.
In the hetero-methylene zone of the 13C NMR spectrum we found the three signals at δ 75.3, 68.5 and 52.3 ppm, similar to those reported.15 In order to confirm these data and according to the DEPT(135) experiment we found that the signals at δ 75.3 and δ 52.3 ppm are negative peaks, and therefore can be assigned to CH2–O and CH2–N signals, whereas the signal at δ 68.5 ppm is positive and can be assigned to the CH–O moiety. Accordingly, the presence of one CH2–O, one CH2–N, and one CH–O group is compatible with 5a. Three years later Liu and Zhou et al.14 committed the same mistake with the incorrect structure w4a14 based on the same spectroscopic data already reported.15 Following a similar reasoning for 4a, we found two negative CH2–O signals at δ 66.1 ppm and δ 63.0 ppm, and one positive (CH–N) at δ 51.4 ppm. This structure was also confirmed with HSQC and HMBC experiments to unambiguously establish the structures 4a and 5a (Fig. 3).
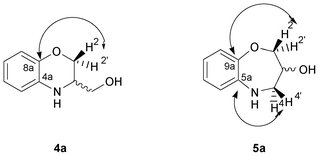 | ||
| Fig. 3 Representation of the HMBC interactions observed in the six- and seven-membered rings (4a and 5a). | ||
In the HMBC experiment to three bonds, the interaction between the hydrogen atoms of the geminal H-2 and H-2′ with the quaternary C-8a of the benzene ring should be observed in 4a, whilst interactions between H-2 [δ 4.25 (ddd)] and H-2′ [δ 3.86 (dd)], and H-4 [δ 3.36 (ddd)] and H-4′ [δ 3.17 (dd)], with the quaternary C-9a (δ 150.7 ppm) and C-5a (δ 142.0 ppm) respectively, are unequivocal proofs for the structure of 5a (Fig. 3).
Both NMR data and the structure of the alcohol obtained fit in with 5a but not with the reported results published.14–16 To reinforce our conclusions even more, we made a deep bibliographic search and found a patent, written in Japanese, in which the synthesis and 1H NMR data of 4a were correctly reported.20
Having found controversial results between our research and that published,14–16 we decided to carry out the reduction of 6 under the conditions previously reported (Fe/HOAc, EtOH/H2O at reflux).15 This procedure provides 5a and not 4a as stated, which we confirmed after an in-depth NMR study and the unambiguous demonstration of the 5a structure, in spite of the fact that it was published as w4a.14–16 Compounds w4b,c were prepared according to a reported procedure,15 and we found that these compounds were actually 5b,c based on DEPT(135) data: 5b and 5c show peaks at δ 52.10 and at δ 52.13 ppm (N–CH2), respectively and negative peaks at δ 75.12 ppm and at δ 75.18 ppm (O–CH2) while the positive peaks at δ 68.30 ppm (O–CH) for both 5b and 5c are unambiguously assigned to the methine carbons. Table 1 shows the correct chemical shifts, multiplicity patterns and coupling constants (J) in Hz of compounds 5a–c and 4a.15
|
|
||||||
|---|---|---|---|---|---|---|
| Compd | R | H-2 | H-2′ | H-3 | H-4 | H-4′ |
| 5a | H | 4.25 (ddd) | 3.86 (dd) | 3.93 (m) | 3.36 (ddd) | 3.17 (dd) |
| J = 12.3, 3.8, 1.4 | J = 12.3, 2.0 | J = 12.9, 4.9, 1.4 | J = 12.9, 2.4 | |||
| 4a | H | 4.18 (dd) | 4.06 (dd) | 3.55 (m) | 3.72 (dd) | 3.64 (dd) |
| J = 10.8, 2.9 | J = 10.8, 5.9 | J = 10.7, 4.9 | J = 10.7, 7.1 | |||
| w4a | H | 4.26 (dd) | 3.88 (dd) | 3.91–3.97 (m) | 3.36 (dd) | 3.19 (dd) |
| J = 12.8, 3.8 | J = 12.8, 2.0 | J = 12.9, 4.8 | J = 12.9, 2.4 | |||
| w4b | 7-Me | 4.25 (dd) | 3.80(dd) | 3.89–3.95 (m) | 3.37 (dd) | 3.14 (dd) |
| J = 12.3, 2.7 | J = 12.3, 1.5 | J = 12.8, 3.9 | J = 12.8, 1.9 | |||
| w4c | 7-tBu | 4.25 (dd) | 3.84 (dd) | 3.89–3.97 (m) | 3.39 (dd) | 3.18 (dd) |
| J = 12.3, 3.7 | J = 12.3, 1.9 | J = 12.8, 4.7 | J = 12.8, 2.2 | |||
In spite of the fact that Liu and Zhou14 reported the incorrect structure of w4a, the formation of six- and/or seven-membered alcohols starting from di-or three-substituted epoxides is accurate. In these cases 2D NMR data correctly supported the target structures and the aromatic splitting patterns comply with our above-mentioned rules.
Conclusively and to emphasize the reactivity of the seven-membered secondary alcohol 5a, its reaction with several purines under the microwave-mediated Mitsunobu conditions was studied (Scheme 3).
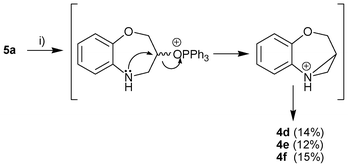 | ||
| Scheme 3 Reagents and conditions: substituted purines, Ph3P, DIAD, anhydrous THF, microwave irradiation, 140 °C, 5 min. | ||
The product of the three reactions are the six-membered compounds 4d–f (3,4-dihydro-2H-3-hydroxymethyl-1,4-benzoxazines linked to the N-9′ purine atom through a methylene linker), demonstrated by the 2D NMR data. The ring contraction can be explained through an aziridinium ion, as previously published in a similar reaction via an episulfonium intermediate.3 The anticancer activity and study of the mechanism of action of 4d–f are underway.
In summary, 2,3,4,5-tetrahydro-1,5-benzoxazepines-3-ol 5a–c have not been reported yet because they were mistaken as being the 3,4-dihydro-2H-3-hydroxymethyl-1,4-benzoxazines 4a–c.14 We have also improved the method of synthesis of 4a and 5a, in a one-step and one-pot reaction from o-aminophenol and epichlorohydrin under microwave irradiation. Compounds 5a–c may serve as synthons for the development of new biologically active prototypes. Although six-membered rings are more stable than their seven-membered counterparts, and in general favored when there are both possibilities of formation, the use of the DEPT, HSQC and HMBC techniques would have made clear the correct assignment of the target scaffold.
We wish to help people working on benzo-fused six- and seven-membered diheterocycles to facilitate their research after having made clear and unambiguous the differences between these two scaffolds. Clarification of the confusion reported in previous works will prevent the proliferation of scientific mistakes and consequently avoid a negative domino effect due to the rapid divulgation of information nowadays.
Experimental section
Melting points were taken in open capillaries on an electrothermal melting point apparatus and are uncorrected. Analytical thin layer chromatography was performed using Merck Kieselgel 60 F254 aluminum sheets, the spots being developed with UV light (λ = 254 nm). Evaporations were carried out in vacuo with a Büchi rotary evaporator and the pressure was controlled by a Vacuubrand CVCII apparatus. For flash chromatography, Merck silica gel 60 with a particle size of 0.040–0.063 mm (230–400 mesh ASTM) was used. Nuclear magnetic resonance spectra have been carried out at the Centro de Instrumentación Científica/Universidad de Granada, and recorded on a 500 MHz 1H and 125 MHz 13C NMR DEPT, HMBC and HSQC NMR Varian NMR-System-TM400 (compounds 4d–f,5a) or 400 MHz 1H and 100 MHz 13CNMR and DEPT NMR Varian Inova-TM spectrometers (compounds 6–8) at ambient temperature. Chemical shifts (δ) are quoted in parts per million (ppm) and are referenced to the residual solvent peak. Signals are designated as follows: s, singlet; d, doublet; dd, doublet of doublets; ddd, double doublet of doublets; dt, doublet of triplets; t, triplet; m, multiplet. The HMBC spectra were measured using a pulse sequence optimized for 10 Hz (inter-pulse delay for the evolution of long-range couplings: 50 ms) and a 5/3/4 gradient combination. In this way, direct responses (J couplings) were not completely removed. High-resolution nano-assisted laser desorption/ionization (NALDI-TOF) or electrospray ionization (ESITOF) mass spectra were carried out on a Bruker Autoflex or a Waters LCT Premier Mass Spectrometer, respectively. Small scale microwave-assisted synthesis was carried out in an Initiator 2.0 single-mode microwave instrument producing controlled irradiation at 2.450 GHz (Biotage AB, Uppsala). Reaction time refers to hold time at 140 °C, not to total irradiation time. The temperature was measured with an IR sensor outside the reaction vessel. Anhydrous THF was purchased from VWR International Eurolab. Anhydrous conditions were performed under argon. 6-Chloropurine, 6-bromopurine and 2,6-dichloropurine were purchased from Aldrich. Compounds 4d–f were synthesized according to our previously reported procedure.3 An HPLC apparatus and a fluorescence detector were used for separation of 4a and 5a (semi-preparative column C-18, 60 Å, 6 μm , 17.8 mm × 300 mm).1,2-Epoxy-(o-nitrophenoxy)propane (6) and 1-(o-nitrophenoxy)-3-chloro-2-propanol (7)
1,2-Epoxy-(o-nitrophenoxy)propane (6) from 1-(o-nitrophenoxy)-3-chloro-2-propanol (7)
To a solution of 7 (2.175 g, 9.3 mmol) in anhydrous THF (30 mL) under argon, 60% NaH (0.373 g, 9.3 mmol) was added dropwise at room temperature. The reaction changed instantaneously from a yellow to a cloudy orange color. 2N HCl was added at pH = 6–7, the mixture was extracted (H2O and CH2Cl2), the organic layers decanted, dried (Na2SO4) and filtered to give 6 (1.33 g, 73%, Scheme 1).1,2-Epoxy-(o-nitrophenoxy)propane (6), 1-(o-nitrophenoxy)-3-chloro-2-propanol (7) and 1,3-bis(o-nitrophenoxy)-2-propanol (8) under microwave irradiation
Epichlorohydrin was added to a solution of o-nitrophenol and aqueous NaOH. The reaction was irradiated in a microwave oven at 95 °C for 40 min. The mixture was cooled and poured into ice water, and the aqueous layer was extracted with CH2Cl2. Organic layers were combined and dried (Na2SO4). After filtration, the solvent was removed in vacuo and the crude was purified by gradient-elution flash column chromatography using hexane/EtOAc (9/1 → 8/2 → 0/10) mixtures.Method b): preparation of 8
Same conditions as method a), except that 2.5 equiv. of NaOH were used and following the general method, 8 (83%) was obtained as a yellowish syrup. 1H NMR (400 MHz, CDCl3) δ 7.82 (d, J = 8.1 Hz, 1H), 7.50 (t, J = 7.5 Hz, 1H), 7.07 (d, J = 8.3 Hz, 1H), 7.00 (t, J = 7.7, 1H), 4.14 (m, 3H), 3.79 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 152.3, 139.6, 134.9, 126.1, 121.1, 115.0, 71.3, 70.0. HR TOF MS ES+, calcd for C15H14N2O7Na (M + Na)+ 357.0699, found 357.0694. Anal. Calcd for C15H14N2O7: C, 53.89; H, 4.22; N, 8.38. Found: C, 53.97; H, 4.12; N, 8.61.2,3,4,5-Tetrahydro-1,5-benzoxazepine-3-ol (5a) and 3,4-dihydro-2H-3-hydroxymethyl-1,4-benzoxazine (4a)
6-Chloro-9-(2,3-dihydro-1,4-benzoxazine-3-ylmethyl)-9H-purine (4d)
Yellowish solid (14%), (mp 77–78 °C), 1H NMR (500 MHz, CDCl3) δ 8.74 (s, 1H), 8.06 (s, 1H), 6.82 (m, 2H), 6.70 (td, J = 7.8, 1.5 Hz, 1H), 6.58 (dd, J = 7.8, 1.5 Hz, 1H), 4.49 (dd, J = 14.1, 5.2 Hz, 1H), 4.36 (dd, J = 14.1, 8.2 Hz, 1H), 4.13 (ddd, J = 25.6, 11.1, 2.7 Hz, 2H), 4.03 (m, 1H), 3.96 (s, 1H). 13C NMR (125 MHz, CDCl3) δ 152.36, 152.20, 151.64, 146.29, 143.52, 132.08, 131.15, 122.78, 119.80, 117.55, 116.33, 65.44, 49.46, 46.53. HR TOF MS ES+, calcd for C14H13ClN5O (M + H)+ 302.0808, found 302.0808. Anal. Calcd for C14H12ClN5O: C, 55.73; H, 4.01; N, 23.21. Found: C, 55.70; H, 4.05; N, 23.50.6-Bromo-9-(2,3-dihydro-1,4-benzoxazine-3-ylmethyl)-9H-purine (4e)
Yellowish solid (12%), (mp 72–73 °C), 1H NMR (500 MHz, CDCl3) δ 8.70 (s, 1H), 8.07 (s, 1H), 6.82 (m, 2H), 6.71 (td, J = 7.8, 1.5 Hz, 1H), 6.58 (dd, J = 7.8, 1.5 Hz, 1H), 4.49 (dd, J = 14.1, 5.2 Hz, 1H), 4.35 (dd, J = 14.1, 8.3 Hz, 1H), 4.13 (ddd, J = 26.6, 11.1, 2.7 Hz, 2H), 4.02 (m, 1H), 3.90 (s, 1H). 13C NMR (125 MHz, CDCl3) δ 152.21, 150.84, 150.71, 146.05, 143.69, 134.60, 131.00, 122.69, 119.75, 117.48, 116.27, 65.33, 49.37, 46.46. HR TOF MS ES+, calcd for C14H13BrN5O (M + H)+ 346.0303, found 346.0305. Anal. Calcd for C14H12BrN5O: C, 48.57; H, 3.49; N, 20.23. Found: C, 48.61; H, 3.55; N, 20.00.2,6-Dichloro-9-(2,3-dihydro-1,4-benzoxazine-3-ylmethyl)-9H-purine (4f)
White solid (15%), (mp 89–90 °C), 1H NMR (500 MHz, CDCl3) δ 8.03 (s, 1H), 6.82 (m, 2H), 6.71 (td, J = 7.7, 1.5 Hz, 1H), 6.59 (dd, J = 7.8, 1.5 Hz, 1H), 4.46 (dd, J = 14.1, 5.2 Hz, 1H), 4.32 (dd, J = 14.1, 8.3 Hz, 1H), 4.17 (dd, J = 11.2, 2.5 Hz, 1H), 4.08 (dd, J = 11.2, 2.6 Hz, 1H), 4.00 (s, 1H), 3.88 (s, 1H). 13C NMR (125 MHz, CDCl3) δ 153.44, 153.33, 152.26, 146.91, 143.41, 131.13, 130.78, 122.77, 119.87, 117.53, 116.39, 65.20, 49.25, 46.45. HR TOF MS ES+, calcd for C14H12Cl2N5O (M + H)+ 336.0419, found 336.0419. Anal. Calcd for C14H11Cl2N5O: C, 50.02; H, 3.30; N, 20.83. Found: C, 49.90; H, 3.35; N, 20.91.Acknowledgements
This study was supported by the Instituto de Salud Carlos III (FIS) through project no. PI10/00592. The M.E-G.R. FPU grant AP2007-02954 is greatly acknowledged.References
- G. Guillaumet, A. R. Katritzky, C. W. Rees and E. F. V. Scriven. Editors (1st ed.), Comprehensive Heterocyclic Chemistry II, Pergamon, Oxford, 19966p. 447 Search PubMed.
- O. Cruz-López, M. C. Núñez, A. Conejo-García, M. Kimatrai and J. M. Campos, Curr. Org. Chem., 2011, 15, 869 CrossRef.
- M. Díaz-Gavilán, A. Conejo-García, O. Cruz-López, M. C. Núñez, D. Choquesillo-Lazarte, J. M. González-Pérez, F. Rodríguez-Serrano, J. A. Marchal, A. Aránega, M. A. Gallo, A. Espinosa and J. M. Campos, ChemMedChem, 2008, 3, 127 CrossRef.
- A. Conejo-García, M. E. García-Rubiño, J. A. Marchal, M. C. Núñez, A. Ramírez, S. Cimino, M. A. García, A. Aránega, M. A. Gallo and J. M. Campos, Eur. J. Med. Chem., 2011, 46, 3795 CrossRef.
- Y. Liao, B. J. Venhuis, N. Rodenhuis, W. Timmerman, H. Wikstrom, E. Meire, G. D. Bartoszyk, H. Bottcher, C. A. Seyfried and S. Sundell, J. Med. Chem., 1999, 42, 2235 CrossRef CAS.
- M. Díaz-Gavilán, J. A. Gómez-Vidal, F. Rodríguez-Serrano, J. A. Marchal, O. Caba, A. Aránega, M. A. Gallo, A. Espinosa and J. M. Campos, Bioorg. Med. Chem. Lett., 2008, 18, 1457 CrossRef.
- M. Iwahashi, E. Takahashi, M. Tanaka, Y. Matsunaga, Y. Okada, R. Matsumoto, F. Nambu, H. Nakai and M. Toda, Bioorg. Med. Chem., 2011, 19, 5361 CrossRef CAS.
- A. N. Matralis, M. G. Katselou, A. Nikitakis and A. P. Kourounakis, J. Med. Chem., 2011, 54, 5583 CrossRef CAS.
- N. Siddiqui, R. Ali, M. S. Alam and W. J. Ahsan, Chem. Pharm. Res., 2010, 2, 309 CAS.
- M. Akhter, S. Habibullah, S. M. Hasan, M. M. Alam, N. Akhter and M. Shaquiquzzaman, Med. Chem. Res., 2011, 20, 1147 CrossRef CAS.
- L. Wang, H. Ankati, S. K. Akubathini, M. Balderamos, Ch. A. Storey, A. V. Patel, V. Price, D. Kretzschmar, E. R. Biehl and S. R. D'Mello, J. Neurosci. Res., 2010, 88, 1970 CAS.
- E. N. Koini, P. Papazafiri, A. Vassilopoulos, M. Koufaki, Z. Horvath, I. Koncz, L. Virag, G. J. Papp, A. Varro and Th. Calogeropoulou, J. Med. Chem., 2009, 52, 2328 CrossRef CAS.
- M. Díaz-Gavilán, D. Choquesillo-Lazarte and J. M. Campos, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o2940 Search PubMed.
- Q.-y. Meng, Q. Liu, J. Li, R.-G. Xing, X.-X. Shen and B. Zhou, Synlett, 2009, 3283 CAS.
- P.-F. Jiao, B.-X. Zhao, W.-W. Wang, Q.-X. He, M.-Sh. Wan, D.-S. Shin and J.-Y. Miao, Bioorg. Med. Chem. Lett., 2006, 16, 2862 CrossRef CAS.
- B. Zhao, S. Zhang and P. Jiao, CN 1847232 A, Appl. No. CN 2006-10043245, March 21st, 2006.
- J. E. Baldwin, J. Chem. Soc., Chem. Commun., 1976, 734 RSC.
- Y. Xu, J.-H. Xu, J. Pan, L. Zhao and S.-L. Zhang, J. Mol. Catal. B: Enzym., 2004, 27, 155 CrossRef CAS.
- B. M. Khadilkar and P. M. Bendale, Synth. Commun., 1997, 27, 2051 CrossRef.
- K. Hamamura, T. Oda, M. Kusaka and N. Kanzaki, WO 2005019188 A1 20050303, 2005; Appl. No. WO 2004-JP12322, August 20th, 2004.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of the 1H, 13C NMR, and DEPT spectra of 4a,d–f, 5a, 6, 7, and 8, HSQC and HMBC charts for 4a,d–f and 5a are provided. See DOI: 10.1039/c2ra21706f |
| This journal is © The Royal Society of Chemistry 2012 |