Catalytic conversion of biomass-derived carbohydrates into fuels and chemicals via furanic aldehydes
Lei
Hu
a,
Geng
Zhao
a,
Weiwei
Hao
a,
Xing
Tang
a,
Yong
Sun
a,
Lu
Lin
*a and
Shijie
Liu
b
aSchool of Energy Research, Xiamen University, Xiamen, 361005, China. E-mail: lulin@xmu.edu.cn; Fax: +86-0592-5952786; Tel: +86-0592-5952786
bDepartment of Paper and Bioprocess Engineering, State University of New York, College of Environmental Science and Forestry, Syracuse, 13210, NY, USA
First published on 24th August 2012
Abstract
In recent years, substantial interest has been devoted to the conversion of biomass-derived carbohydrates into furanic aldehydes such as furfural, 5-hydroxymethylfurfural (HMF) and 5-halomethylfurfural, because these products are considered as important versatile intermediates that can be further transformed into a wide variety of high performance fuels and high value-added chemicals. However, low yields and high production costs that are due to the special chemical structures and properties of biomass-derived carbohydrates to a large extent have limited the practical production of furanic aldehydes. Recently, various catalytic conversion strategies have been developed to overcome these limitations. In this review, we systematically summarize and discuss catalytic conversion strategies from the perspective of catalysts and reaction solvents as well as formation mechanisms and influencing factors for the production of furanic aldehydes from biomass-derived carbohydrates. Meanwhile, we also outline the synthesis of furanic aldehyde-based fuels such as 2-methylfuran (MF), 2,5-dimethylfuran (DMF), 5-ethoxymethylfurfural (EMF) and alkanes and chemicals such as levulinic acid (LA), 2,5-diformylfuran (DFF) and 2,5-furandicarboxylic acid (FDCA). Moreover, some potential research orientations are proposed based on the major problems encountered in recent research.
 Lei Hu | Lei Hu is a PhD candidate at School of Energy Research, Xiamen University, China. In September 2010 he joined in Biomass Chemical Catalytic Conversion Engineering Laboratory. His research interests are in the field of catalytic conversion of renewable biomass into fuels and chemicals via furanic aldehydes. |
 Yong Sun | Yong Sun earned his PhD degree in 2008 from South China University of Technology, Guangzhou, China. He is an assistant professor at School of Energy Research, Xiamen University, China. He has been engaged in research work on Biomass Chemical Engineering over seven years and has authored and co-authored over thirty publications in the area of structure of lignin and cellulose, pretreatment and chemical transformation of renewable biomass. |
 Lu Lin | Lu Lin received his PhD degree in 1994 from Sun Yat-sen University, Guangzhou, China. He served as senior visiting scholar from October 1998 to January 2000 in North Carolina State University, United States. He is interested in the production of biofuels and high-value chemicals from lignocellulosic biomass. He has published more than one hundred peer-review papers and two monographs. From 2007, he has served on BioResources as one of co-editors and Journal of Bio-Based Materials and Bioenergy and Journal of Bioprocess Engineering and Biorefinery as one of editors. In addition, he is also the laureate of five prizes awarded by the Ministry of Education of China and the other related institution for his active achievements in recent years. |
 Shijie Liu | Shijie Liu obtained his PhD degree in 1992 from University of Alberta, Edmonton, Canada. He has been a professor in the SUNY ESF department of Paper and Bioprocess Engineering (PBE) since 2005. His research interests include chemical and biochemical kinetics, process modeling and optimization, fluid particle systems, fiber properties, and mass transfer. His research is widely published with numerous book chapters and articles in journals of chemical engineering. He holds leadership roles in several professional engineering societies. He currently is active for Journal of Bio-Based Materials and Bioenergy as one of executive editors and Journal of Bioprocess Engineering and Biorefinery as Editor-in-Chief. |
1. Introduction
With growing concerns about diminishing fossil resources, global warming and environmental pollution, the search for renewable resources has attracted worldwide attention.1–6 In this respect, biomass, which is widespread, abundant and inexpensive, is regarded to be an ideal substitute for fossil resources.7–12 Nature produces 170 billion metric tons of biomass per year by photosynthesis, 75% of which can be assigned to carbohydrates.13–16 As a result, it is of great importance to look for efficient approaches to utilize biomass-derived carbohydrates. Nowadays, one of the most attractive and promising approaches is to convert biomass-derived carbohydrates into furanic aldehydes such as furfural, 5-hydroxymethylfurfural (HMF) and 5-halomethylfurfural including 5-chloromethylfurfural (CMF) and 5-bromomethylfurfural (BMF), which have been recognized as important versatile platform compounds for the synthesis of a broad range of new products as well as for the replacement of fossil resources-derived fuels and chemicals (Scheme 1).17–21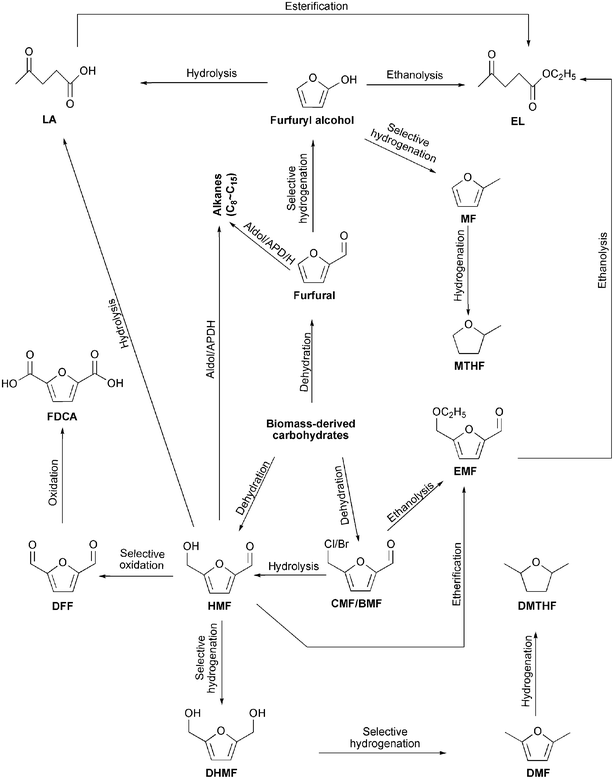 | ||
| Scheme 1 Furanic aldehydes as platform compounds for the production of fuels and chemicals. | ||
Furfural, which is the dehydration product of xylose22–25 and can be converted to furfuryl alcohol, 2-methylfuran (MF),26 2-methyltetrahydrofuran (MTHF),20,26 linear alkanes,27,28 phenol-formaldehyde resin,29 furoic acid and maleic acid,13,20,30 has been produced on an industrial scale for decades. Currently, approximately 70% of the global furfural production is concentrated in China.13,18,31 HMF, which is formed from hexose,32–38 was first reported at the end of the 19th century. It is a more multifunctional molecule than furfural because it is simultaneously an aromatic aldehyde, an aromatic alcohol and a furan ring system. Thus, HMF has been called a “sleeping giant”5,13,15 and used for the production of 2,5-dihydroxymethylfuran (DHMF),39 2,5-dimethylfuran (DMF),39 2,5-dimethyltetrahydrofuran (DMTHF),40 2,5-diformylfuran (DFF),41 2,5-furandicarboxylic acid (FDCA),42 levulinic acid (LA)43 and linear alkanes.44 More recently, Mascal et al.45–48 and Kumari et al.49 reported two new platform chemicals, CMF and BMF, respectively. Similar to furfural and HMF, CMF and BMF can be subsequently transformed into valuable fuels and chemicals such as 2,5-dimethylfuran (DMF), 5-ethoxymethylfurfural (EMF) or LA45–49. However, to the best of our knowledge, the production of HMF, CMF and BMF has not yet reached industrial scale because of low yields and high production costs. In the past few years, many researchers have endeavored to explore some new high-efficiency and low-cost processes for the conversion of biomass-derived carbohydrates into furfural, HMF, CMF and BMF. In 2008, Mamman et al.31 reviewed technologies and processes used for the production of furfural based on acid-catalyzed hydrolysis of hemicellulose. In 2011, Karinen et al.18 and Zakrzewska et al.15 summarized achievements in ionic liquid-mediated and heterogeneous-catalyzed formation of HMF, respectively. However, the synthesis of CMF and BMF were not mentioned. In addition, this area has been progressing very fast. Hence, a comprehensive review is also needed.
In this review, the state-of-the-art of catalytic conversion approaches for the production of furfural, HMF, CMF and BMF from biomass-derived carbohydrates is systematically summarized and discussed, focusing on catalysts, reaction solvents, formation mechanisms and influencing factors. Moreover, some emphasis is also put on the synthesis of furfural-, HMF-, CMF- and BMF-based derivatives such as MF, DMF, EMF, linear alkanes, LA, DFF and FDCA. Furthermore, we also point out some potential research trends in the further studies.
2. Production of furanic aldehydes
The industrial application of furanic aldehydes largely depends on the high-effective and cost-competitive production of furanic aldehydes from biomass-derived carbohydrates. Compared to conventional catalytic conversion systems, cheaper materials, new-style catalysts and solvents have been gradually used for furanic aldehydes production. The detailed advancements are reviewed below.2.1. Furfural production
2.1.1. Xylose and xylan as feedstock
Furfural is typically derived from xylose that is mainly present as xylan in the hemicellulose.50 The traditional processes for the production of furfural were based on homogeneous acid catalysts such as HCOOH, CH3COOH, HCl, H2SO4, HNO3 and H3PO4 in aqueous solution.31,51–57 However, these homogeneous acid catalysts are very corrosive and possess higher environmental risks.20,23,31 Recently, numerous modifications based on the use of solid acids, Lewis acids and various solvents have been proposed to design a cleaner and more environmental friendly process (Table 1). For example, O'Neill et al.58 studied the dehydration of xylose in water by the use of H-ZSM-5 catalyst, 46% furfural yield was obtained at 200 °C over 18 min. Dhepe and Sahu59 reported a one-pot conversion of hemicellulose into furfural using K10 and HUSY in aqueous media, gave 12% yields at 170 °C for 3 h, respectively. In addition, Sn-beta, MSHS-SO3H, graphene, graphene oxide (GO), sulfonated grapheme (SG) and sulfonated graphene oxide (SGO) were also synthesized and used for furfural formation, and yields of 14.3, 43.5, 51, 53, 55 and 62% were achieved, respectively.23,60,61 It is worth noting that water is the most economical solvent for the synthesis of furfural. However, it can accelerate some of the undesired side reactions that decrease the yields of furfural.18 Thus, polar aprotic solvents were subsequently introduced to the production of furfural. Takagaki et al.62 showed that a furfural yield of 37% was obtained from xylose in N,N-dimethylformamide (DMF) using Amberlyst-15 with addition of hydrotalcite (HT). When the dehydration of xylose was performed in dimethyl sulfoxide (DMSO) by the use of Nafion 117, 60% furfural yield was achieved.63 Although polar aprotic solvents inhibit the formation of side-products to some extent, they also suffer from the drawback of the poor solubility of carbohydrates and the problem of high boiling points, which are unfavorable to the production and separation of furfural.18,50| Substrate | Solvent | Catalyst | T/°C | t/min | Yield (%) | Ref. |
|---|---|---|---|---|---|---|
| Xylose | H2O | HCl | 170 | 50 | 30 | 54 |
| Xylose | H2O | HNO3 | 180 | 20 | 3.5 | 56 |
| Xylose | H2O | H3PO4 | 180 | 20 | 27.6 | 56 |
| Xylose | H2O | H2SO4 | 135 | 90 | 17.3 | 55 |
| Xylose | H2O | HCOOH | 180 | 20 | 23.8 | 56 |
| Xylose | H2O | CH3COOH | 180 | 20 | 15.8 | 56 |
| Xylose | H2O | GO | 200 | 35 | 53 | 23 |
| Xylose | H2O | SG | 200 | 35 | 55 | 23 |
| Xylose | H2O | SGO | 200 | 35 | 62 | 23 |
| Xylose | H2O | Graphene | 200 | 35 | 51 | 23 |
| Xylose | H2O | H-ZSM-5 (28) | 200 | 18 | 46 | 58 |
| Xylose | H2O | Sn-beta (100) | 110 | 180 | 14.3 | 60 |
| Xylose | H2O | MSHS-SO3H | 190 | 60 | 43.5 | 61 |
| Xylose | ACN | HT-Amberlyst-15 | 100 | 180 | 37 | 62 |
| Xylose | DMF | HT-Amberlyst-15 | 100 | 180 | 37 | 62 |
| Xylose | DMSO | MCM-41 | 140 | 1440 | 44.7 | 66 |
| Xylose | DMSO | Nafion 117 | 150 | 120 | 60 | 63 |
| Xylose | DMSO | Amberlyst-15 | 170 | 240 | 78 | 66 |
| Xylose | DMSO | H-mordenite (20) | 140 | 240 | 24 | 70 |
| Xylose | DMSO | H3PW12O40/MCM-41 | 140 | 240 | 52 | 68 |
| Xylose | DMSO | Cs3PW12O40/MCM-41 | 140 | 240 | 45 | 69 |
| Xylose | H2O/MIBK | HCl | 170 | 60 | 85 | 54 |
| Xylose | H2O/MIBK | HY-faujasite (15) | 170 | 50 | 29.7 | 64 |
| Xylose | H2O/MIBK | H-mordenite (12) | 170 | 50 | 20.2 | 64 |
| Xylose | H2O/MIBK | MCM-41-SO3H | 140 | 1440 | 51 | 66 |
| Xylose | H2O/toluene | ITQ-2 | 170 | 384 | 66 | 78 |
| Xylose | H2O/toluene | (VO)2P2O7 | 170 | 360 | 56 | 74 |
| Xylose | H2O/toluene | SAPO-11 | 180 | 240 | 50.9 | 72 |
| Xylose | H2O/toluene | SO42−/SnO2 | 100 | 2880 | 31.1 | 76 |
| Xylose | H2O/toluene | BEA/TUD-1 | 170 | 480 | 74 | 78 |
| Xylose | H2O/toluene | Amberlyst 70 | 175 | 200 | 70 | 65 |
| Xylose | H2O/toluene | SBA-15-SO3H | 160 | 240 | 68.3 | 75 |
| Xylose | H2O/toluene | Del-Nu-6(1) (29) | 170 | 240 | 47 | 71 |
| Xylose | H2O/toluene | Hβ-zeolite (25) | 140 | 240 | 39.8 | 70 |
| Xylose | H2O/toluene | HY-zeolite (5.1) | 140 | 240 | 41.1 | 70 |
| Xylose | H2O/toluene | HY-faujasite (15) | 170 | 50 | 41.8 | 64 |
| Xylose | H2O/toluene | H-mordenite (13) | 260 | 3 | 98 | 73 |
| Xylose | H2O/toluene | H4TiNbO5-MgO | 160 | 240 | 55 | 67 |
| Xylose | H2O/toluene | H-MCM-22 (24) | 170 | 384 | 71 | 78 |
| Xylose | H2O/toluene | MCM-41-SO3H | 140 | 1440 | 76 | 66 |
| Xylose | H2O/toluene | H-Al-MCM-41 (12) | 160 | 360 | 50 | 71 |
| Xylose | H2O/toluene | H3PW12O40/MCM-41 | 160 | 240 | 48 | 68 |
| Xylose | H2O/toluene | Cs3PW12O40/MCM-41 | 160 | 240 | 33 | 69 |
| Xylose | H2O/toluene | SO42−/ZrO2–Al2O3/SBA-15 | 160 | 240 | 52.7 | 24 |
| Xylose | H2O/1-butanol | MCM-41 | 170 | 240 | 44.1 | 25 |
| Xylose | (H2O–DMSO)/DCM | HCl | 140 | 180 | 56.9 | 52 |
| Xylose | (H2O–DMSO)/(2-butanol–MIBK) | HCl | 140 | 12 | 64.6 | 52 |
| Xylose | H2O–acetone–DMSO | SO42−/C | 230 | 12 | 11.5 | 77 |
| Xylose | [EMIM][HSO4] | [EMIM][HSO4] | 100 | 30 | 62 | 79 |
| Xylose | [SBMIM][HSO4] | [SBMIM][HSO4] | 150 | 25 | 91.5 | 80 |
| Xylose | DMA–[EMIM]Cl | CrCl2 | 100 | 120 | 45 | 22 |
| Xylose | DMA–[EMIM]Cl | CrCl3 | 100 | 120 | 40 | 22 |
| Xylose | DMA–[BMIM]Br | CrCl2 | 100 | 240 | 55 | 22 |
| Xylose | [BMIM]Cl/toluene | H2SO4 | 100 | 240 | 44 | 79 |
| Xylose | [EMIM][HSO4]/toluene | [EMIM][HSO4] | 100 | 360 | 84 | 79 |
| Xylan | H2O | CH3COOH | 180 | 20 | 22 | 56 |
| Xylan | H2O–acetone–DMSO | SO42−/C | 230 | 12 | 8 | 77 |
| Xylan | [EMIM]Cl | CrCl2–HCl | 140 | 120 | 25 | 22 |
| Xylan | [BMIM]Cl | CrCl3 | 200 | 2 | 63 | 81 |
| Xylan | [EMIM][HSO4]/toluene | [EMIM][HSO4] | 100 | 240 | 29 | 79 |
| Hemicellulose | H2O | K10 | 170 | 180 | 12 | 59 |
| Hemicellulose | H2O | HUSY | 170 | 180 | 12 | 59 |
| Hemicellulose | H2O | H2SO4 | 170 | 60 | 10 | 59 |
In 1998, mixed solvents consisting of water and toluene or methyl isobutyl ketone (MIBK) in the presence of H-form faujasites and mordenites were found to be effective for the conversion of xylose into furfural.64 In biphasic systems, the dehydration reaction takes place in the aqueous phase and furfural is extracted to the organic phase as soon as it is formed,64 which contributes to reduce several unwanted secondary reactions and improve furfural yields. Hence, the biphasic systems were further studied by a number of research groups that used various solid acids including ion-exchange resins, zeolites, sulfated metal oxides and supported heteropoly acids, furfural yields ranging from 20.2 to 98% were obtained.24,25,65–78 Solid acids are an attractive option for the synthesis of furfural because they are less corrosive and can be easily separated and reused.18 However, solid acids may be gradually deactivated due to the deposition of humins and the loss of active sites after a few reaction cycles, and consequently, require frequent regeneration by calcination and impregnation,50 respectively.
In recent years, ionic liquids, which possess some specific properties such as low melting point, negligible vapor pressure, non-flammability, high thermal stability, remarkable solubilizing ability and close to infinite structural variation,13,15 have been successfully used for the production of furfural from xylose and xylan. Lima et al.79 and Tao et al.80 found that acidic ionic liquids 1-ethyl-3-methylimidazolium hydrogen sulfate ([EMIM][HSO4]) and 1-(4-sulfonic acid)butyl-3-methylimidazolium hydrogen sulfate ([SBMIM][HSO4]) as both solvents and catalysts were effective for the conversion of xylose into furfural, and yields of 84 and 91.5% could be reached, respectively. Neutral ionic liquid 1-butyl-3-methylimidazolium chloride ([BMIM]Cl) as solvent was also active when H2SO4 was used as catalyst.79 Binder et al.22 investigated the conversion of xylose in the presence of N,N-dimethylacetamide (DMA) using CrCl2 as catalyst, the results indicated that the addition of 1-butyl-3-methylimidazolium bromide ([BMIM]Br) obviously improved the yield of furfural. When xylan was used as substrate by the use of CrCl2 and HCl, a furfural yield of 25% was achieved in [EMIM]Cl.22 In addition, Zhang and Zhao81 demonstrated that xylan was effectively converted into furfural in [BMIM]Cl with addition of CrCl3 under microwave irradiation, and furfural yield could be up to 63%. Although good results were obtained, it should be pointed out that the long-term recyclability of ionic liquids is critical for the development of cost-effective technologies based on ionic liquids because they are very expensive. Furthermore, the separation and purification of furfural from ionic liquids have not yet been fully demonstrated.
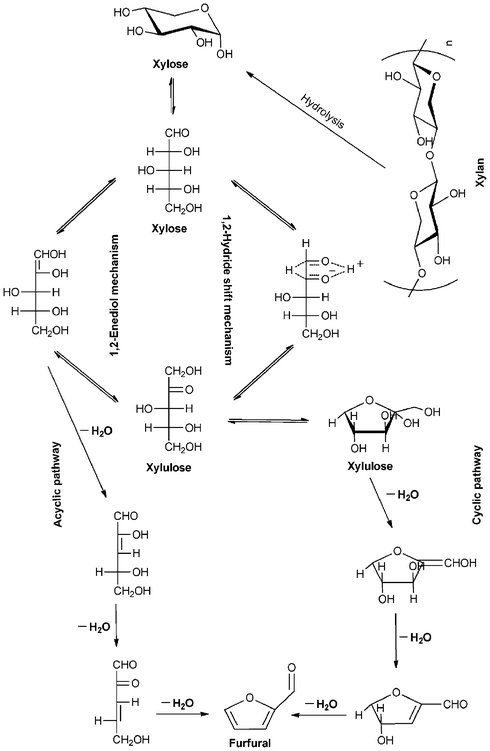 | ||
| Scheme 2 Proposed mechanisms for the production of furfural from xylose. Adapted from refs. 22, 60 and 83. | ||
2.2. HMF production
| Substrate | Catalyst | Solvent | T/°C | t/min | Yield (%) | Ref. |
|---|---|---|---|---|---|---|
| Fructose | HCl | H2O | 180 | 3 | 24.2 | 39 |
| Fructose | HCl | [BMIM]Cl | 80 | 8 | 97 | 90 |
| Fructose | HCl | tert-butanol | 120 | 120 | 70 | 93 |
| Fructose | HCl | Isopropanol | 120 | 120 | 83 | 93 |
| Fructose | HCl | H2O/THF | 150 | 35 | 56.4 | 94 |
| Fructose | HCl | H2O/MIBK | 180 | 3 | 54.6 | 35 |
| Fructose | HCl | H2O/1-butanol | 180 | 15 | 53.1 | 94 |
| Fructose | HCl | H2O/1-hexanol | 180 | 3 | 32 | 39 |
| Fructose | HCl | H2O/(2-butanol–toluene) | 180 | 3 | 49.9 | 39 |
| Fructose | HCl | (H2O–DMSO)/DCM | 140 | 120 | 87 | 52 |
| Fructose | HCl | (H2O–DMSO–PVP)/(2-butanol–MIBK) | 180 | 3 | 68.1 | 35 |
| Fructose | HBr | Sulfolane | 100 | 60 | 93 | 89 |
| Fructose | HNO3 | [BMIM]Cl | 80 | 5 | 93 | 90 |
| Fructose | H2SO4 | Isopropanol | 120 | 120 | 57 | 93 |
| Fructose | H2SO4 | [BMIM]Cl | 100 | 50 | 82.9 | 88 |
| Fructose | H2SO4 | (H2O–DMSO)/(2-butanol–MIBK) | 180 | 3 | 60.4 | 35 |
| Fructose | H3PO4 | [BMIM]Cl | 80 | 720 | 67 | 90 |
| Fructose | H3PO4 | (H2O–DMSO)/(2-butanol–MIBK) | 180 | 3 | 38.8 | 35 |
| Fructose | HCOOH | H2O | 200 | 10 | 58 | 91 |
| Fructose | CH3COOH | H2O | 200 | 20 | 58 | 91 |
| Fructose | CH3COOH | [BMIM]Cl | 80 | 720 | 78 | 90 |
| Fructose | Maleic acid | [BMIM]Cl | 80 | 50 | 88 | 90 |
| Fructose | Nafion-H | DMSO | 120 | 120 | 75 | 99 |
| Fructose | Amberlyst-15 | [BMIM]Cl | 80 | 10 | 83.3 | 100 |
| Fructose | Amberlyst-15 | Isopropanol | 120 | 240 | 45 | 93 |
| Fructose | Amberlyst-15 | THF | 120 | 20 | 48 | 101 |
| Fructose | Amberlyst-15 | DMSO | 120 | 240 | 100 | 99 |
| Fructose | Amberlyst-15 | H2O/MIBK | 115 | 60 | 47.8 | 112 |
| Fructose | Dowex 50wx8-100 | H2O–acetone | 150 | 15 | 73.4 | 97 |
| Fructose | Dowex 50wx8-100 | Acetone–DMSO | 150 | 20 | 89.8 | 98 |
| Fructose | H-mordenite (11) | H2O/MIBK | 165 | 90 | 74 | 102 |
| Fructose | H-ZSM-5 (13) | H2O/MIBK | 165 | 300 | 45 | 103 |
| Fructose | Hβ-zeolite (15.6) | H2O/MIBK | 165 | 300 | 32 | 103 |
| Fructose | SBA-15-SO3H | [BMIM]Cl | 120 | 60 | 81 | 104 |
| Fructose | TiO2 | H2O | 200 | 5 | 38.1 | 105 |
| Fructose | ZrO2 | H2O | 200 | 5 | 30.5 | 105 |
| Fructose | CeO2–Nb2O5 | H2O | 130 | 360 | — | 106 |
| Fructose | SO42−/ZrO2 | [BMIM]Cl | 100 | 30 | 88.4 | 107 |
| Fructose | SO42−/ZrO2 | Acetone–DMSO | 180 | 20 | 61.2 | 10 |
| Fructose | SO42−/ZrO2 | H2O/MIBK | 115 | 60 | 49.2 | 112 |
| Fructose | SO42−/C | Acetone–DMSO | 200 | 12 | 27 | 77 |
| Fructose | TiO2 nanoparticles | H2O | 120 | 5 | 34.3 | 108 |
| Fructose | TiO2 nanoparticles | DMSO | 140 | 5 | 54.1 | 108 |
| Fructose | Nb2O5 | H2O/2-butanol | 160 | 100 | 46 | 109 |
| Fructose | Ta2O5 | H2O/2-butanol | 160 | 100 | 62 | 109 |
| Fructose | TA-p | H2O/2-butanol | 160 | 100 | 90 | 109 |
| Fructose | P2O5 | [BMIM]Cl | 50 | 60 | 81.2 | 110 |
| Fructose | H3PW12O40 | [EMIM]Cl | 80 | 60 | 80 | 113 |
| Fructose | FePW12O40 | DMSO | 120 | 120 | 49 | 99 |
| Fructose | Ag3PW12O40 | H2O/MIBK | 120 | 60 | 77.7 | 111 |
| Fructose | Cs2.5H0.5PW12O40 | H2O/MIBK | 115 | 60 | 74 | 112 |
| Fructose | [MIMPS]3PW12O40 | 2-Butanol | 120 | 120 | 99.1 | 114 |
| Fructose | H3PW12O40/MIL-101 | DMSO | 130 | 30 | 63 | 113 |
| Fructose | H3PW12O40/MIL-101 | [EMIM]Cl | 80 | 150 | 79 | 113 |
| Fructose | IrCl3 | [BMIM]Cl | 120 | 20 | 89.2 | 119 |
| Fructose | CrCl2 | ChCl | 100 | 30 | 40 | 115 |
| Fructose | CrCl3 | ChCl | 100 | 30 | 60 | 115 |
| Fructose | CrCl3 | DMSO | 130 | 180 | 71.1 | 116 |
| Fructose | CrCl3 | [BMIM]Cl | 100 | 1 | 78 | 34 |
| Fructose | AlCl3 | H2O | 120 | 5 | 53.9 | 117 |
| Fructose | AlCl3 | DMSO | 140 | 5 | 71.3 | 117 |
| Fructose | AlCl3 | H2O/MIBK | 130 | 5 | 61.2 | 117 |
| Fructose | GeCl4 | [BMIM]Cl | 100 | 5 | 92.1 | 121 |
| Fructose | WCl6 | [BMIM]Cl | 50 | 240 | 63 | 124 |
| Fructose | WCl6 | [BMIM]Cl/THF | 50 | 240 | 72 | 124 |
| Fructose | NH4Cl | Isopropanol | 120 | 720 | 68.2 | 123 |
| Fructose | Sc(OTf)3 | DMSO | 120 | 120 | 83.3 | 122 |
| Fructose | Yb(OTf)3 | DMSO | 120 | 120 | 80.2 | 122 |
| Fructose | Ho(OTf)3 | DMSO | 120 | 120 | 78.1 | 122 |
| Fructose | Sm(OTf)3 | DMSO | 120 | 120 | 73 | 122 |
| Fructose | Nd(OTf)3 | DMSO | 120 | 120 | 63.5 | 122 |
| Fructose | CrCl2/NHC | DMSO | 100 | 360 | 52 | 37 |
| Fructose | CrCl2/NHC | [BMIM]Cl | 100 | 360 | 95 | 37 |
| Fructose | FeCl3–Et4NBr | DMSO | 90 | 180 | 53 | 118 |
| Fructose | FeCl3–Et4NBr | DMF | 90 | 120 | 62 | 118 |
| Fructose | FeCl3–Et4NBr | NMP | 90 | 120 | 86 | 118 |
| Fructose | [HMIM]Cl | [HMIM]Cl | 90 | 45 | 92 | 125 |
| Fructose | ChCl–citric acid | (ChCl–citric acid)/ethyl acetate | 80 | 60 | 91 | 126 |
| Fructose | [EMIM][HSO4] | [EMIM][HSO4]/toluene | 100 | 30 | 79 | 79 |
| Fructose | [EMIM][HSO4] | [EMIM][HSO4]/MIBK | 100 | 30 | 88 | 79 |
| Fructose | [BMIM][HSO4] | [BMIM]Cl | 80 | 30 | 80 | 90 |
| Fructose | [SBMIM][HSO4] | [BMIM]Cl | 80 | 26 | 91 | 90 |
| Fructose | [NMP][CH3SO3] | DMSO | 120 | 120 | 69.4 | 128 |
| Fructose | [NMP][HSO4] | DMSO | 120 | 120 | 72.3 | 128 |
| Fructose | [DiEG(MIM)2][OMs]2 | [DiEG(MIM)2][OMs]2 | 120 | 40 | 69.8 | 129 |
| Fructose | [TriEG(MIM)2][OMs]2 | [TriEG(MIM)2][OMs]2 | 120 | 40 | 77.2 | 129 |
| Fructose | [TetraEG(MIM)2][OMs]2 | [TetraEG(MIM)2][OMs]2 | 120 | 40 | 92.3 | 129 |
| Fructose | ILIS-SO3H | DMSO | 100 | 4 | 70.1 | 130 |
| Fructose | ILIS-SO2Cl | DMSO | 100 | 4 | 67.2 | 130 |
| Fructose | ILISNP-HSO4 | DMSO | 130 | 30 | 63 | 131 |
| Fructose | No catalyst | H2O–DMSO | — | 80 | 85 | 132 |
| Fructose | No catalyst | (BHC–ChCl)/MIBK | 110 | 60 | 84 | 36 |
| Fructose | No catalyst | [BMIM]Cl | 155 | 1 | 98 | 133 |
| Fructose | No catalyst | [BEMIM)Cl | 140 | 50 | 53.2 | 88 |
| Fructose | No catalyst | [HexMIM]Cl | 140 | 50 | 21.9 | 88 |
| inulin | HCl | (H2O–DMSO)/DCM | 140 | 150 | 70 | 52 |
| Inulin | HCl | [OMIM]Cl/ethyl acetate | 120 | 60 | 50.9 | 95 |
| Inulin | HCl | (H2O–DMSO)/(2-butanol–MIBK) | 170 | 5 | 75.5 | 52 |
| Inulin | TA-p | H2O/2-butanol | 160 | 150 | 87 | 109 |
| Inulin | CrCl2 | ChCl | 90 | 60 | 36 | 115 |
| Inulin | CrCl3 | ChCl | 90 | 60 | 46 | 115 |
| Inulin | AlCl3 | H2O | 120 | 5 | 29.4 | 117 |
| Inulin | AlCl3 | DMSO | 140 | 5 | 39.2 | 117 |
| Inulin | AlCl3 | H2O/MIBK | 130 | 5 | 32.5 | 117 |
| Inulin | [EMIM][HSO4] | [EMIM][HSO4]/MIBK | 100 | 30 | 73 | 79 |
| Inulin | ChCl–oxalic acid | ChCl–oxalic acid | 80 | 120 | 56 | 127 |
| Inulin | ChCl–oxalic acid | (ChCl–oxalic acid)/ethyl acetate | 80 | 120 | 64 | 127 |
| Inulin | No catalyst | BHC–ChCl–H2O | 110 | 60 | 52 | 36 |
Qi et al.97,98 studied the dehydration of fructose in the presence of a strong acidic resin Dowex 50wx8-100 in water–acetone and acetone–DMSO, HMF yields of 73.4 and 89.8% were achieved, respectively. Nafion-H was used for the dehydration of fructose in DMSO, HMF yield was 75%. When the reaction was performed under mild evacuation, the yield of HMF could be improved to 94%.99 Subsequently, another acidic resin Amberlyst-15 was also tested for the formation of HMF from fructose in [BMIM]Cl, and a yield of 83.3% was reached.100 In addition, HMF yields with 45 and 48% were obtained from fructose by the use of Amberlyst-15 when isopropyl alcohol and tetrahydrofuran (THF) were used as reaction solvents, respectively.93,101 Surprisingly, an extraordinary HMF yield of 100% was achieved in DMSO when Amberlyst-15 was reduced to powder in a size of 0.15–0.053 mm.99 The authors proposed that a decrease in the size of Amberlyst-15 enhances the removal of adsorbed water from the surface and near-surface of the catalyst.99 To the best of our knowledge, this is the highest yield of HMF from fructose to date.
Moreau et al.102 examined the application of H-form mordenite for the dehydration of fructose and obtained 74% HMF yield in water/MIBK. When H-ZSM-5 and Hβ-zeolite were used in the same solvent, HMF yields of 45 and 32% were achieved, respectively.103 Recently, SBA-15-SO3H was also synthesized and used for the dehydration of fructose in [BMIM]Cl, 81% HMF yield was obtained.104 Moreover, the evaluation of these catalysts indicated that the acidity and pore size of zeolites have a large effect on catalytic activities in the synthesis of HMF.102–104
In 2008, Qi et al.105 reported the formation of HMF from fructose by the use of TiO2 and ZrO2 in water, gave 38.1 and 30.5% HMF yields, respectively. Subsequently, they found that SO42−/ZrO2 was also effective in acetone–DMSO and [BMIM]Cl, 72.8 and 88.4% HMF yields could be achieved, respectively.10,106 Recently, mixed oxide CeO2–Nb2O5, sulfonated carbon-based catalyst (SO42−/C) and self-assembled mesoporous TiO2 nanoparticles (NPs) were prepared, and displayed excellent catalytic activities for the production of HMF from fructose.77,107,108 Yang109 found that Nb2O5 and Ta2O5 also possessed good performance on fructose dehydration and the latter was more effective than the former. More importantly, H3PO4–modified Ta2O5 (TA-p) exhibited stronger acidity, which is highly favorable for the synthesis of HMF, and a yield of 90% was obtained in water/2-butanol. When inulin was used as feedstock, 87% HMF yield was achieved. Surprisingly, Ray et al.110 found that a high HMF yield of 81.2% was reached when P2O5 was utilized for the dehydration of fructose in [BMIM]Cl in 60 min at a lower temperature of 50 °C, which can be due to acidic nature and hygroscopic property of P2O5.
12-Phosphotungstic acid (H3PW12O40), which is a heteropolyacid and is classified as a superacid with strong acidity, has been used as catalyst for the production of HMF.99 However, it is soluble in water and polar solvents, which limits its application. In 2011, two solid heteropolyacid salts Cs2.5H0.5PW12O40 and Ag3PW12O40 were prepared and tested for the dehydration of fructose, HMF yields of 74 and 77.7% were observed in the presence of water/MIBK, respectively.111,112 In the same year, H3PW12O40 was encapsulated into a chromium-based metal-organic framework (MIL-101) and used as a solid acid catalyst for the dehydration of fructose, and a yield of HMF with 79% was achieved in [EMIM]Cl. When DMSO was used as solvent, HMF yield was 63%.113 In addition, the results also indicated that good catalytic performance of H3PW12O40/MIL-101 can be attributed to some protons released from H3PW12O40 in MIL-101.113 Currently, a salt of H3PW12O40 containing ionic liquid-based cation ([MIMPS]3PW12O40) was synthesized and employed for the synthesis of HMF from fructose, a yield of 99.1% was reached in the case of 2-butanol.114 This should be the first example of the application of ionic liquid-based heteropolyacid salt in carbohydrate dehydration.
In 2007, Zhao et al.38 found that many Lewis acids are very effective to catalyze fructose into HMF. When CrCl2, CrCl3, FeCl2, CuCl2, VCl3, MoCl3, PdCl2, PtCl4, RuCl3 and RhCl3 were used as catalysts, HMF yields ranging from 63 to 83% were achieved in the presence of [EMIM]Cl. Subsequently, CrCl2 and CrCl3 were further studied in DMSO, ChCl and [BMIM]Cl, which gave good HMF yields from fructose and inulin.34,37,115,116 AlCl3 is known for HMF synthesis from fructose in [BMIM]Cl.38 However, high yields of HMF of 53.9, 61.2 and 71.3% were also reached when water, water/MIBK and DMSO were used as solvents, respectively.117 When inulin was utilized as feedstock in the same reaction conditions, HMF yields of 29.4, 32.5 and 39.2% were obtained, respectively.117 Tong et al.118 found that a combination of FeCl3 and tetraethylammonium bromide (Et4NBr) as catalysts were effective for the dehydration of fructose, 53, 62 and 86% HMF yields were observed in the presence of DMSO, DMF and NMP, respectively. Meanwhile, FeCl3Br− ion was considered as the active catalyst species. Recently, novel Lewis acids such as IrCl3 and GeCl4 were also found to be very active for the dehydration of fructose in [BMIM]Cl, excellent yields of HMF of 89.2 and 92.1% were achieved, respectively.119–121 Wang et al.122 showed that rare earth metal trifluoromethanesulfonates including Sc(OTf)3, Yb(OTf)3, Ho(OTf)3, Sm(OTf)3 and Nd(OTf)3 as new type of Lewis-acid catalysts exhibited high catalytic activities that increased with decreasing ionic radius of rare-earth metal cations. A maximum HMF yield of 83.3% was observed in the presence of DMSO by using Sc(OTf)3. More interestingly, Liu et al.123 demonstrated that HMF could be obtained in good yield of 68.2% using NH4Cl for the dehydration of fructose in isopropanol. At the same time, isopropanol showed better selectivity of HMF than methanol and ethanol. Although excellent HMF yields were reached using the above mentioned Lewis acid catalysts for the dehydration of fructose, these catalytic systems required temperatures of ≥80 °C. Surprisingly, good HMF yield of 63% could be obtained at a lower temperature of 50 °C in 4 h when WCl6 was used as catalyst in the presence of [BMIM]Cl. The yield of HMF could be improved to 72% when using continuous extraction with THF.124
The dehydration of fructose into HMF can be catalyzed by acidic ionic liquids in different solvents. In a pioneering work, Moreau et al.125 investigated the synthesis of HMF from fructose using 1-H-3-methyl imidazolium chloride ([HMIM]Cl) as catalyst and solvent, a yield as high as 92% was achieved. Hu et al.126 obtained 91% HMF yield from fructose in the mixture of ChCl and citric acid using ethyl acetate as extraction solvent. When inulin was used as substrate, the yield of HMF was 64% in the same catalytic system.127 When [EMIM][HSO4] was used with MIBK for the dehydration of fructose, this led to 88% HMF yields. In the same reaction conditions, 73% HMF yield was achieved when inulin was utilized as substrate.79 In addition, 1-butyl-3-methylimidazolium bisulfate ([BMIM][HSO4]), [SBMIM][HSO4], N-methyl-2-pyrrolidonium methyl sulfonate ([NMP][CH3SO3]) and N-methyl-2-pyrrolidonium hydrogen sulfate ([NMP][HSO4]) also showed high catalytic activities in the dehydration of fructose, HMF yields of 80, 91, 69.4 and 72.3% were obtained, respectively.90,128 Presently, dicationic acidic ionic liquids were found to be active for the formation of HMF from fructose, yields of 69.8, 77.2 and 92.3% were achieved when diethylene glycol-bis(3-methylimidazolium) dimesylate ([DiEG(MIM)]2[OMs]2), triethylene glycol-bis(3-methylimidazolium) dimesylate ([TriEG(MIM)]2[OMs]2) and tetraethyleneglycol-bis(3-methylimidazolium) dimesylate ([TetraEG(MIM)]2[OMs]2) were employed as catalysts and solvents, respectively.129 Moreover, it is reported that Lewis-acidic ionic liquids were more effective than Brønsted-acidic ionic liquids for the production of HMF from fructose in the same reaction conditions.130 More interestingly, Bao et al.130 and Sidhpuria et al.131 reported that 3-allyl-1-(4-sulfobutyl)imidazolium trifluromethanesulfonate, 3-allyl-1-(4-sulfurylchloridebutyl)imidazolium trifluromethanesulfonate and 1-(triethoxysilylpropyl)-3-methylimidazolium hydrogen sulfate could be immobilized on silica gel or silica nanoparticles (ILIS-SO3H, ILIS-SO2Cl and ILISNP-HSO4) and applied for the dehydration of fructose into HMF, yields of 70.1, 67.2 and 63% were observed in DMSO, respectively. Furthermore, these results also demonstrated that supported acidic ionic liquid catalysts required smaller amounts of ionic liquids and simultaneously minimized limitations associated to their viscosity, separation, corrosiveness and toxicity.131
In addition to the various catalysts mentioned above, fructose can also be dehydrated into HMF in the absence of catalysts. A new method for the synthesis of HMF from fructose was presented by using constant-current electrolysis, led to 85% yield at room temperature in DMSO with traces of water, which is ascribed to in situ electrogenerated acid (EGA).132 Vigier et al.136 showed that betaine hydrochloride (BHC)-based media could be used for the dehydration of fructose into HMF. In particular, a yield of 84% was obtained in a biphasic system of BHC–ChCl/MIBK. When inulin was used as substrate in the same reaction conditions, 52% HMF yield was achieved in BHC–ChCl–H2O. Moreover, high HMF yields ranging from 21.9 to 98% were observed in the presence of [BMIM]Cl, 1-benzyl-3-methylimidazolium chloride ([BEMIM]Cl) and 1-hexyl-3-methylimidazolium chloride ([HexMIM]Cl), which can be due to the acidity of the C-2 hydrogen in imidazolium ring and chloride ion (Cl−) as a base and a nucleophile.88,133,134
| Substrate | Catalyst | Solvent | T/°C | t/min | Yield (%) | Ref. |
|---|---|---|---|---|---|---|
| Glucose | HCl | H2O | 170 | 45 | 2.2 | 52 |
| Glucose | HCl | H2O–DMSO | 170 | 10 | 10.7 | 52 |
| Glucose | HCl | (H2O–DMSO)/DCM | 140 | 270 | 29.8 | 52 |
| Glucose | HCl | (H2O–DMSO)/(2-butanol–MIBK) | 170 | 10 | 22.8 | 52 |
| Glucose | H2SO4 | (H2O–DMSO)/(2-butanol–MIBK) | 170 | 60 | 16.3 | 52 |
| Glucose | H3PO4 | (H2O–DMSO)/(2-butanol–MIBK) | 170 | 60 | 17.3 | 52 |
| Glucose | [NMP][HSO4] | DMSO | 90 | 120 | 2.4 | 136 |
| Glucose | [NMP][CH3SO3] | DMSO | 90 | 120 | 3 | 136 |
| Glucose | H3PW12O40/MIL-101 | [EMIM]Cl | 100 | 180 | 2 | 113 |
| Glucose | CrCl2 | [EMIM]Cl | 100 | 180 | 70 | 38 |
| Glucose | CrCl2/NHC | [BMIM]Cl | 100 | 360 | 81 | 37 |
| Glucose | CrCl3 | ChCl | 110 | 30 | 31 | 115 |
| Glucose | CrCl3 | TEAC | 130 | 10 | 71.3 | 139 |
| Glucose | CrCl3 | TBAC | 110 | 240 | 49 | 140 |
| Glucose | CrCl3 | CPL–LiCl | 100 | 180 | 66.7 | 138 |
| Glucose | CrCl3 | [EMIM]Cl | 90 | 180 | 72 | 137 |
| Glucose | CrCl3 | [BMIM]Cl | 120 | 240 | 81 | 79 |
| Glucose | CrCl3 | DMA–LiCl | 120 | 120 | 55 | 7 |
| Glucose | CrCl3 | TBAC–DMA | 110 | 240 | 52 | 140 |
| Glucose | CrCl3 | [BMIM]Cl/MIBK | 120 | 240 | 79 | 79 |
| Glucose | CrCl3 | [BMIM]Cl/toluene | 120 | 240 | 91 | 79 |
| Glucose | CrCl3/NHC | [BMIM]Cl | 100 | 360 | 81 | 37 |
| Glucose | SnCl4 | CPL–LiCl | 100 | 180 | 64.7 | 138 |
| Glucose | SnCl4 | [EMIM][BF4] | 100 | 180 | 61 | 33 |
| Glucose | AlCl3 | H2O | 120 | 20 | 40.3 | 117 |
| Glucose | AlCl3 | DMSO | 120 | 5 | 52.4 | 117 |
| Glucose | AlCl3 | H2O/THF | 160 | 10 | 52 | 141 |
| Glucose | AlCl3 | H2O/MIBK | 130 | 5 | 43 | 117 |
| Glucose | IrCl3 | [BMIM]Cl | 140 | 30 | 7.5 | 119 |
| Glucose | GeCl4 | [BMIM]Cl | 120 | 30 | 47.5 | 121 |
| Glucose | ScCl3 | DMA | 200 | 7.5 | 30 | 143 |
| Glucose | YbCl3 | [BMIM]Cl | 200 | 5 | 26 | 144 |
| Glucose | TiO2 | H2O | 200 | 5 | 18.6 | 105 |
| Glucose | TiO2 nanoparticles | H2O | 120 | 5 | 24.8 | 108 |
| Glucose | TiO2 nanoparticles | NMP | 140 | 5 | 29.6 | 108 |
| Glucose | TiO2 nanoparticles | DMSO | 140 | 5 | 37.2 | 108 |
| Glucose | TiO2 nanoparticles | H2O/MIBK | 130 | 5 | 25.9 | 108 |
| Glucose | ZrO2 | H2O | 200 | 5 | 10 | 105 |
| Glucose | ZrO2 | [BMIM]Cl–H2O | 200 | 10 | 53 | 16 |
| Glucose | SO42−/ZrO2 | DMSO | 130 | 240 | 19.2 | 135 |
| Glucose | SO42−/ZrO2–Al2O3 | DMSO | 130 | 240 | 47.6 | 135 |
| Glucose | NA-p | H2O | 120 | 180 | 52.1 | 145 |
| Glucose | NA-p | H2O/2-butanol | 160 | 110 | 49 | 146 |
| Glucose | H-ZSM-5 | [BMIM]Cl | 110 | 480 | 45 | 140 |
| Glucose | Ag3PW12O40 | H2O/MIBK | 130 | 240 | 76.3 | 111 |
| Glucose | Titanate nanotube | H2O | 120 | 180 | 14.5 | 147 |
| Glucose | CrCl2-Im-SBA-15 | (H2O–DMSO)/(2-butanol–MIBK) | 150 | 180 | 35 | 148 |
| Glucose | LCC | DMSO–[BMIM]Cl | 160 | 50 | 68 | 149 |
| Glucose | HT-Amberlyst-15 | DMF | 80 | 540 | 42.3 | 151 |
| Glucose | HT-Amberlyst-15 | DMF | 100 | 180 | 42.3 | 152 |
| Glucose | Sn-Beta-HCl | H2O/THF | 180 | 70 | 56.9 | 153 |
| Glucose | AlCl3–HCl | SBP | 170 | 35 | 60.9 | 154 |
| Glucose | AlCl3–HCl | LDS | 170 | 35 | 48.4 | 154 |
| Glucose | Isomerase-HCl | H2O/1-butanol | 190 | 45 | 63.3 | 155 |
| Glucose | B2O3 | [BMIM]Cl | 120 | 300 | 60 | 157 |
| Glucose | B(OH)3 | H2O/MIBK | 150 | 300 | 14 | 32 |
| Glucose | B(OH)3 | [EMIM]Cl | 120 | 180 | 42 | 156 |
| Sucrose | [HMIM]Cl | [HMIM]Cl | 90 | 30 | — | 125 |
| Sucrose | [NMM][CH3SO3] | DMA–LiBr | 90 | 120 | 47.5 | 136 |
| Sucrose | [TriEG(MIM)2][OMs]2 | [TriEG(MIM)2][OMs]2 | 120 | 150 | 61.2 | 129 |
| Sucrose | HCl | (H2O–DMSO)/DCM | 140 | 270 | 50.8 | 52 |
| Sucrose | HCl | (H2O–DMSO)/(2-butanol–MIBK) | 170 | 5 | 50.1 | 52 |
| Sucrose | HT-Amberlyst-15 | DMF | 120 | 180 | 53.9 | 151 |
| Sucrose | AlCl3 | DMSO | 140 | 5 | 42.5 | 117 |
| Sucrose | CrCl2 | ChCl | 100 | 60 | 62 | 115 |
| Sucrose | CrCl3 | ChCl | 100 | 60 | 43 | 115 |
| Sucrose | CrCl3 | [BMIM]Cl | 100 | 5 | 76 | 34 |
| Sucrose | CrCl3 | [BMIM]Cl/MIBK | 100 | 240 | 100 | 79 |
| Sucrose | GeCl4 | [BMIM]Cl | 120 | 30 | 55.4 | 121 |
| Sucrose | SnCl4 | [EMIM][BF4] | 100 | 180 | 65 | 33 |
| Sucrose | ZnCl2 | [OMIM]Cl | 120 | 60 | 68.4 | 158 |
| Sucrose | CrCl2–HCl | [OMIM]Cl | 120 | 30 | 82 | 158 |
| Sucrose | FeCl3-Et4NBr | DMF | 90 | 180 | 38 | 118 |
| Sucrose | FeCl3-Et4NBr | NMP | 90 | 180 | 40 | 118 |
| Sucrose | FeCl3-Et4NBr | DMSO | 90 | 300 | 27 | 118 |
| Sucrose | CrCl3-NH4Br | DMA | 100 | 60 | 87 | 159 |
| Cellobiose | HCl | (H2O–DMSO)/DCM | 140 | 570 | 38.3 | 52 |
| Cellobiose | HCl | (H2O–DMSO)/(2-butanol–MIBK) | 170 | 10 | 27 | 52 |
| Cellobiose | AlCl3 | H2O/THF | 160 | 10 | 58 | 141 |
| Cellobiose | CrCl3 | [BMIM]Cl | 140 | 5 | 55 | 34 |
| Cellobiose | CrCl3 | [BMIM]Cl/MIBK | 100 | 240 | 50 | 79 |
| Cellobiose | SnCl4 | [EMIM][BF4] | 100 | 180 | 57 | 33 |
| Cellobiose | GeCl4 | [BMIM]Cl | 120 | 30 | 41 | 121 |
| Cellobiose | HT-Amberlyst-15 | DMF | 120 | 180 | 38.4 | 151 |
| Cellulose | CrCl2 | DMA–LiCl | 140 | 120 | 54 | 7 |
| Cellulose | CrCl2 | [EMIM]Cl–H2O | 120 | 360 | 89 | 160 |
| Cellulose | CrCl3 | [EMIM]Cl | 200 | 2.5 | 62 | 81 |
| Cellulose | CrCl2–CuCl2 | [EMIM]Cl | 120 | 480 | 55.4 | 161 |
| Cellulose | CrCl2–RuCl3 | [BMIM]Cl | 120 | 180 | 60 | 162 |
| Cellulose | CrCl3–LiCl | [BMIM]Cl | 160 | 10 | 62.3 | 11 |
| Cellulose | CrCl3–Zr(O)Cl2 | DMA–LiCl | 120 | 120 | 62 | 163 |
| Cellulose | Cr[(DS)H2PW12O40]3 | H2O | 150 | 120 | 52.7 | 164 |
A major breakthrough came in 2007 when Zhao et al.38 published their pioneering work for the dehydration of glucose in the presence of [EMIM]Cl using CrCl2 as catalyst, which gave an outstanding HMF yield as high as 70%. Meanwhile, the catalytic activity of CrCl2 was found to be higher than CrCl3, which has been questioned in later studies. Yong et al.37 reported the production of HMF from glucose using NHC/chromium chloride (NHC: N-heterocyclic carbene) in [BMIM]Cl, and HMF yield of 81% was achieved, which exhibited no significant difference between CrCl2 and CrCl3. However, Zhang et al.137 found that CrCl3 was much more active than CrCl2 in the dehydration of glucose into HMF. Compared to CrCl2, CrCl3 has a stronger Lewis acidity and a better solubility in ionic liquids, which are helpful to promote the isomerization of glucose into fructose and accelerate the dehydration of fructose into HMF.137 Moreover, CrCl3 is more stable, easily handled under air and much cheaper, thus, it was chosen to replace CrCl2. In the subsequent studies, CrCl3 was further evaluated in the case of various solvents such as [BMIM]Cl/toluene, [BMIM]Cl/MIBK, ChCl, DMA–LiCl, caprolactam (CPL)–LiCl, tetraethylammonium chloride (TEAC) and tetrabutylammonium chloride (TBAC),7,8,79,115,138–140 HMF yields ranging from 31 to 91% were obtained. Moreover, SnCl4 was found to be an effective catalyst for the dehydration of glucose into HMF. Interestingly, the yield of 64.7% in CPL–LiCl was higher than 61% in 1-ethyl-3-methylimidazolium tetrafluoroborate [EMIM][BF4] in the same reaction conditions.33,138 In addition to CrCl2/3 and SnCl4, a number of other metal chlorides such as AlCl3, GeCl4, MgCl2, ScCl3 and YbCl3 could also be used for the dehydration of glucose into HMF with moderate yields of 40.3–72%.117,121,141–144
The synthesis of HMF from glucose in the presence of solid acid catalysts is now also established. TiO2 and ZrO2 were employed in various solvents including water, DMSO, NMP, water/MIBK and [BMIM]Cl–water for the dehydration of glucose into HMF. The results indicated that TiO2 and ZrO2 could not only promote the isomerization of glucose into fructose acting as base catalysts, but also promote the dehydration of fructose into HMF acting as acid catalysts.16,95,105,108 SO42−/ZrO2 and SO42−/ZrO2–Al2O3 were also prepared and used for the synthesis of HMF from glucose. The results demonstrated that the latter was more effective than the former in DMSO, this might be ascribed to the introduction of Al, which resulted in an increase in basicity that promoted the isomerization of glucose into fructose.135 Nakajima et al.145 and Yang et al.146 investigated the dehydration of glucose into HMF using H3PO4-modified Nb2O5 (NA-p) as catalyst in water and water/2-butanol, yields of 52.1 and 49% were obtained, respectively. Furthermore, the acidic properties and catalytic activity of Nb2O5 were found to be remarkably enhanced by treatment with H3PO4. Moreover, Fan et al.111 and Jadhav et al.140 found that Ag3PW12O40 and H-ZSM-5 were also suitable for the dehydration of glucose into HMF with good yields of 45 and 76.3%, respectively. More recently, some new solid catalysts have been developed. Protonated titanate nanotube exhibited a much higher catalytic performance for the dehydration of glucose into HMF, which was due to large amounts of Brønsted acid sites, Lewis acid sites and large pores of protonated titanate nanotube.147 Degirmenci et al.148 showed that the introduction of CrCl2 into 1-propyl-3-methylimidazolium chloride ([PMIM]Cl) immobilized on the surface of SBA-15 (CrCl2-Im-SBA-15) gave 35% HMF yield from glucose in water–DMSO/2-butanol–MIBK. More interestingly, various biomass-derived carbonaceous catalysts were also synthesized and tested for the dehydration of glucose. It was found that lignin-derived solid acid catalyst (LCC) was the most active because it has an ideal amorphous structure composed of aromatic carbon sheets oriented in a considerably random fashion and a moderate acid density with 1.34 mmol g−1 –SO3H groups, leading to 68% HMF yield in the presence of DMSO–[BMIM]Cl.149 The above described results are nice improvements over previous reports using homogeneous acid catalysts, however, the stability of these solid acid catalysts need to be further improved.
More recently, Ohara et al.,150 Takagaki et al.151 and Tuteja et al.152 reported the dehydration of glucose into HMF using a combination of HT and Amberlyst-15 under mild conditions. In this new strategy, HT was used as the base catalyst for isomerization of glucose to fructose and Amberlyst-15 was used as the acid catalyst for dehydration of fructose to HMF, which gave 42.3% yield in DMF. Subsequently, this concept was further investigated. Sn-Beta coupled with HCl led to 56.9% HMF yield from glucose in water/THF.153 The joint use of AlCl3 and HCl also resulted in 60.9 and 48.4% HMF yields from glucose in water using 2-sec-butylphenol (SBP) and lignin-derived solvent (LDS) containing alkyl-substituted phenolics such as propyl guaiacol (PG) and propyl syringol (PS) as organic phase, respectively.154 More interestingly, Huang et al.155 demonstrated that enzymatic and acid catalysis could also be combined and employed to dehydrate glucose into HMF. In this process, sodium tetraborate-assisted immobilized glucose isomerase was used to convert glucose into fructose with high yield of 87.8%. And then, fructose was dehydrated into HMF by the use of HCl, a yield of 63.3% was achieved in water/1-butanol.
Compared to the above mentioned approaches catalyzed by metal catalysts, three examples of non-metal-catalyzed dehydration of glucose into HMF have been reported up to date. In a pioneering work, B(OH)3 was found to be less efficient for the synthesis of HMF from glucose in water/MIBK.32 However, when B(OH)3 was utilized in [EMIM]Cl, HMF yield of 42% was observed.156 Unexpectedly, B2O3 showed higher activity in HMF formation from glucose, a good yield of 60% was achieved in [BMIM]Cl.157 The difference between B2O3 and B(OH)3 is due to the ability of the former to facilitate dehydration by the trapping of water. Moreover, the reaction of B2O3 with water can form B(OH)3, which is also a promoter.157 In addition, the results also indicated that Cl− was highly necessary for HMF formation. PF6− was found to suppress the reaction, whereas BF4− was compatible with Cl− and led to excellent activity. The mixture of [BMIM]Cl and [EMIM][BF4] has some advantages for practical applications because it is considerably less viscous than pure [BMIM]Cl.157
The conversion of disaccharides such as sucrose consisting of one molecule of glucose and one molecule fructose and cellobiose consisting of two molecules glucose has been explored (Table 3). In the earlier work, Moreau et al.125 studied the conversion of sucrose in [HMIM]Cl at 90 °C. The results showed that fructose moiety was nicely transformed into HMF with high yield (∼90%) whereas only 3% of glucose moiety was converted after 30 min. In the subsequent studies, N-methylmorpholinium methyl sulfonate ([NMM][CH3SO3]) was used for the synthesis of HMF from sucrose, and the yield was 47.5%.136 Chheda et al.52 and Takagaki et al.151 also investigated the conversion of sucrose and cellobiose into HMF using HCl and a combination of HT and Amberlyst-15 in water–DMSO/2-butanol–MIBK and DMF, respectively, but the results were not ideal. Recently, metal chlorides have been used as catalysts for the conversion of sucrose and cellobiose and good results were obtained. For instance, Hu et al.33 found that SnCl4 was effective to convert sucrose and cellobiose into HMF in [EMIM][BF4], and the yields were up to 65 and 57%, respectively. When CrCl3 was utilized as catalyst, 76% sucrose and 55% cellobiose were converted into HMF in [BMIM]Cl, respectively.34 Surprisingly, using the same catalyst, an outstanding HMF yield of 100% from sucrose was observed in the presence of [BMIM]Cl when MIBK was used as organic phase.79 Moreover, sucrose can also be effectively converted into HMF in the case of AlCl3, CrCl2, GeCl4, ZnCl2 and FeCl3–Et4NBr in various solvents.115,117,118,121,158 More recently, a new catalytic system consisting of a metal chloride and an ammonium halide was developed to produce HMF from sucrose under mild conditions. Among the different combinations, CrCl3–NH4Br led to 87% HMF yield in DMA.159 It should be pointed out that this is the highest HMF yield from sucrose in non-ionic liquid solvents up to now.
Cellulose, which is made up of glucose via β-1,4-glycosidic bonds, is the most abundant carbohydrate in nature. The direct transformation of cellulose into HMF involving hydrolysis, isomerization and dehydration is desirable for industrial production of HMF because it can reduce the step of the production of glucose and the corresponding costs, which has been proved to be feasible in ionic liquids under mild conditions (Table 3). Binder and Raines7 reported that 54% HMF yield was observed from cellulose in DMA–LiCl with [EMIM]Cl additive using CrCl2 as catalyst. The yield of HMF was increased to 62% by the use of CrCl3 in [BMIM]Cl under microwave irradiation.81 Remarkably, extraordinary results were obtained by Zhang et al.160 They designed a catalyst-free system that was composed of [EMIM]Cl and H2O for the conversion of cellulose into water-soluble reducing sugars in high total yield up to 97%. The reducing sugars could be further transformed into HMF with a yield of 89% by adding CrCl2. The calculated pKw values of water indicated that the dissociation constant of water in [EMIM]Cl under mild conditions was over three orders of magnitude higher than pure water under ambient conditions. This increase in the water dissociation constant was essential for the efficient conversion of cellulose into HMF.160 In addition, paired metal chlorides have also been employed in ionic liquids for the conversion of cellulose into HMF. In 2009, Su et al.161 reported the formation of HMF with 55.4% yield from cellulose using a combination of CrCl2 and CuCl2 in [EMIM]Cl. Two years later, the joint use of CrCl2 and RuCl3 resulted in 60% HMF yield in [BMIM]Cl.162The most recent publications showed that approximately 62% HMF yield could also be achieved in DMA–LiCl and [BMIM]Cl when CrCl3 coupled with Zr(O)Cl2 and LiCl were used for the conversion of cellulose, respectively.11,163 These above mentioned results indicated that the paired metal chlorides were more effective than individual metal chloride in the same reaction conditions because the addition of non-chromium chlorides facilitated the hydrolysis of cellulose.11,161–163 More interestingly, a Brønsted–Lewis-surfactant-combined heteropolyacid (Cr[(DS)H2PW12O40]3) was prepared and used as a heterogeneous catalyst for the conversion of cellulose into HMF, 52.7% yield was reached in water. This good catalytic activity was attributed to amphiphilic properties and double acidities of Cr[(DS)H2PW12O40]3.164 Specifically, the former, including hydrophilic and hydrophobic properties, could overcome the insolubility of cellulose in water through the interaction between cellulose and catalyst, and the latter containing Brønsted acidity and Lewis acidity could promote the hydrolysis of cellulose into glucose followed by dehydration of glucose to HMF.164
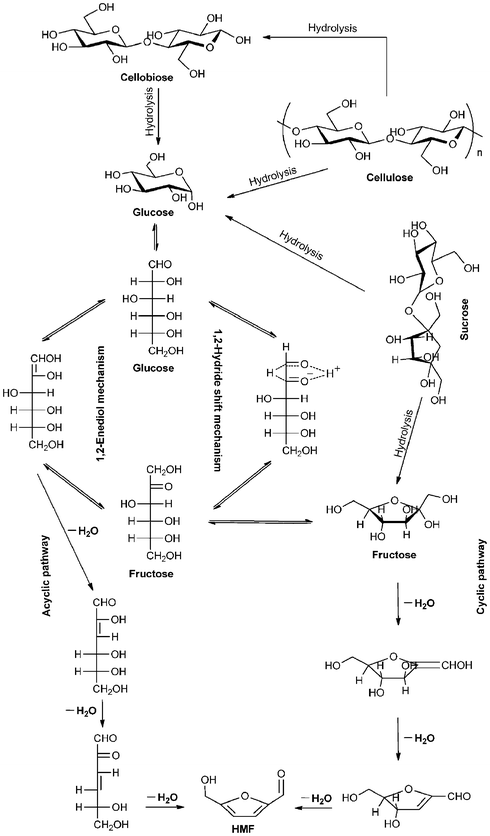 | ||
| Scheme 3 Proposed mechanisms for the production of HMF from glucose. Adapted from refs. 5, 168 and 173. | ||
2.3. CMF and BMF production
| Substrate | Catalyst | Solvent | T/°C | t/min | Yielda (%) | Ref. |
|---|---|---|---|---|---|---|
| a CMF yield; b BMF yield. | ||||||
| Glucose | HCl | HCl/DCE | 100 | 180 | 81.2 | 47 |
| Glucose | HCl | HCl/DCE | 120 | 5 | 58 | 174 |
| Glucose | HCl | HCl/DCM | 100 | 2.5 | 15 | 174 |
| Glucose | HCl | (HCl–LiCl)/DCE | 65 | 1800 | 71 | 45 |
| Glucose | HBr | (HBr–LiBr)/toluene | 65 | 2880 | 50b | 49 |
| Fructose | HCl | HCl/DCM | 100 | 2.5 | 80 | 174 |
| Fructose | HCl | (HCl–LiCl)/DCE | 65 | 1800 | 90 | 49 |
| Fructose | HBr | (HBr–LiBr)/DCE | 25 | 2880 | 68b | 49 |
| Fructose | HBr | (HBr–LiBr)/toluene | 25 | 2880 | 82b | 49 |
| Fructose | HBr | (HBr–LiBr)/THF | 25 | 2880 | 0b | 49 |
| Sucrose | HCl | HCl/DCE | 100 | 180 | 89.7 | 47 |
| Sucrose | HCl | HCl/DCM | 100 | 2.5 | 51 | 174 |
| Sucrose | HCl | (HCl–LiCl)/DCE | 65 | 1800 | 76 | 45 |
| Cellulose | HCl | HCl/DCE | 100 | 180 | 83.5 | 47 |
| Cellulose | HCl | (HCl–LiCl)/DCE | 65 | 1800 | 71 | 45 |
| Cellulose | HBr | (HBr–LiBr)/toluene | 65 | 1680 | 80b | 49 |
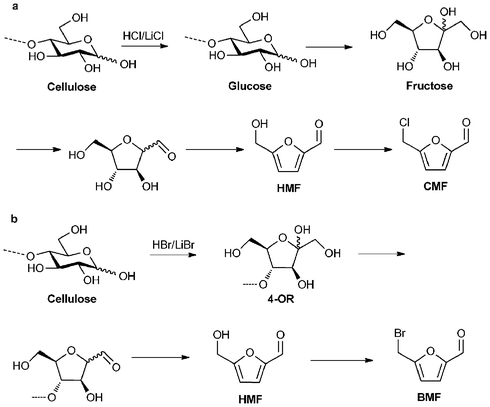 | ||
| Scheme 4 Proposed mechanisms for the production of CMF and BMF from cellulose. Adapted from ref. 49. | ||
3. Influencing factors of production of furanic aldehydes
In the process of the synthesis of furfural, HMF, CMF and BMF, the types of catalysts and solvents are two major parameters to control the dehydration of biomass-derived carbohydrates. However, the catalytic activities strongly depends on other factors such as reaction temperature, reaction time, catalyst loading, substrate concentration, extraction method, heating method, water content, co-solvent and co-catalyst, etc. To better understand the influence of these factors on the catalytic activities and utilize these factors to optimize the reaction conditions for producing furanic aldehydes, these factors should be discussed. In 2011, Zakrzewska et al.15 have detailedly discussed the first five factors. Thus, in this review, only the other four factors will also be systematically discussed.3.1. Heating method
Previously, the production of furanic aldehydes was mainly carried out with conventional heating methods such as an oil-bath and sand-bath. However, these methods, which are based on convective-conductive heating from the surface into the interior of the reaction systems, have several drawbacks such as slow heating rate, high energy-consumption and non-uniform heating. In contrast, microwave irradiation produces internal heating of the reaction systems by direct coupling of microwave energy with solvents, reagents and catalysts, which can overcome the drawbacks of conventional heating methods.175 Thus, in recent years, microwave irradiation has been gradually used for the synthesis of furanic aldehydes. Zhang et al.81 found that 18% furfural yield was produced from pine wood in an oil-bath at 200 °C for 6 min in [BMIM]Cl using CrCl3 as catalyst. While the reaction was performed under microwave irradiation, a yield of furfural with 31% was obtained in 3 min. Qi et al.97 studied the dehydration of fructose in water–acetone by the use of Dowex 50wx8-100. When the reaction was heated by a sand bath, fructose conversion and HMF yield were 22.1 and 13.9% at 150 °C for 10 min, respectively, while the corresponding values under microwave irradiation were increased to 91.7 and 70.3%, respectively. Li et al.176 reported the formation of HMF in yields of 91 and 61% from glucose and cellulose in the presence of CrCl3 under microwave irradiation at 400 W for only 1 and 2 min in [BMIM]Cl, respectively. Whereas, comparison experiments in an oil-bath gave only 17 and 17% HMF yields at 100 °C for 60 and 240 min, respectively. These results also indicated that microwave irradiation not only reduces the reaction time but also improves the conversion of carbohydrates and yields of furanic aldehydes compared to conventional heating methods. With the development of science and technology, large microwave equipment may be applied to the practical production of furanic aldehydes, which will contribute to the large-scale production of biofuels and chemicals.3.2. Water content
In the process of the formation of furanic aldehydes, water plays an important role. On the one hand, water acts as a necessary reactant in the hydrolysis of di/polysaccharides into monosaccharides, on the other hand, it is generally thought to accelerate the decomposition of furanic aldehydes into undesired products.177 As a result, water content is a sensitive issue, especially in non-aqueous systems such as ionic liquids. Recent studies revealed that a small amount of water in [BMIM]Cl, [HexMIM]Cl and TEAC leads to a slight increase in the yields of HMF.16,100,116,139 This may be ascribed to that a small amount of water to some extent not only reduces the viscosity of the ionic liquids, which is beneficial to mass transfer, but also increases the solubility of the substrates and catalysts in the ionic liquids.16,100,116,139 However, when the water content exceeds a certain level, it will result in the precipitation of polysaccharides such as cellulose15,178 and a decrease of HMF yields.16,121,139,156 Thus, it is imperative that water content should be carefully controlled to obtain best catalytic activities.3.3. Co-solvent
In order to accelerate the conversion rate of biomass-derived carbohydrates, improve the selectivity and yield of furanic aldehydes or decrease the production costs by reducing the use of ionic liquids, suitable co-solvents have been introduced into the catalytic systems. For example, Qi et al.97 found that the conversion of fructose and HMF yield were only 57.6 and 32.5% in water using Dowex 50wx8-100 as catalyst, respectively. When 30% acetone was added into water, the corresponding values were increased to 71.9 and 53.1% in the same reaction conditions, respectively. Guo et al.149 reported that fructose conversion and HMF yields were increased from 73 to 96% and from 61 to 82% in the presence of LCC when the weight ratio of DMSO in [BMIM]Cl was increased from 0 to 4/6, respectively. Most remarkably, 99% glucose conversion and 98% HMF selectivity without any humins were achieved using 12-molybdophosphoric acid (H3PMo12O40) as catalyst by adding a certain amount of ACN (about 28.3%) to [EMIM]Cl. In pure [EMIM]Cl, the conversion of glucose and the selectivity of HMF were only 66 and 90% in the same reaction conditions, respectively, whereas the amount of humins was as high as 9%.179 The role of co-solvents in promoting the catalytic activities is not fully understood, however, the available evidences provide several possibilities such as inhibiting the formation of undesired products,97,148,180 decreasing the viscosity of ionic liquids,149,179 reducing the activation energy of related reactions181 and mediating the acidic activity of catalysts.179 Hence, the detailed mechanisms should also be further studied.3.4 Co-catalyst
On the basis of the main catalyst, the addition of co-catalysts can be favorable for the formation of furanic aldehydes. 68.7% furfural yield from xylose was obtained in water using HCl as catalyst. When NaCl was used as co-catalyst, the yield of furfural was increased to 81.3%.86 The authors thought that contrary to what was believed before, the improvement of furfural yield was neither due to the increased activity of the acid, nor to the increased boiling temperature or the salting-out effect, but was due to that NaCl promoted the formation of 1,2-enediol from xylose, which undergoes subsequent acyclic dehydration to furfural.86 When Et4NBr was added in the presence of FeCl3, the yield of HMF from fructose was improved from 42 to 86% in NMP. Based on the experimental data, the authors found that Et4NBr could obviously promote the dehydration of fructose.118 As for glucose, the addition of TEAC based on CrCl2 resulted in an increase of HMF yield from 41.4 to 49.3% in DMSO, which may be attributed to that the presence of TEAC could promote the isomerization of glucose into fructose.116 Moreover, various co-catalysts including NH4Cl, NH4Br, CuCl2, LiCl and RuCl3 added to CrCl2 or CrCl3 were also investigated in the conversion of sucrose and cellulose. The results showed that these co-catalysts could increase the yields of HMF by facilitating the hydrolysis of sucrose and cellulose.11,159–162 As can be seen from the above, co-catalysts can play an important part in each step of the conversion of biomass-derived carbohydrates. Furthermore, the addition of co-catalysts can also reduce the use of expensive catalysts such as CrCl2 to decrease the production costs.4. Synthesis of furanic aldehydes-based derivatives
Numerous furanic aldehydes-based fuels and chemicals have been synthesized through hydrogenation, oxidation, etherification, alcoholysis, aldol condensation or aqueous-phase dehydration/hydrogenation (APD/H) pathways. In the following part, we will mainly review the synthesis of MF, DMF, EMF, C8–C15 linear alkanes, LA, DFF and FDCA from furanic aldehydes or directly from biomass-derived carbohydrates.4.1. Liquid fuels
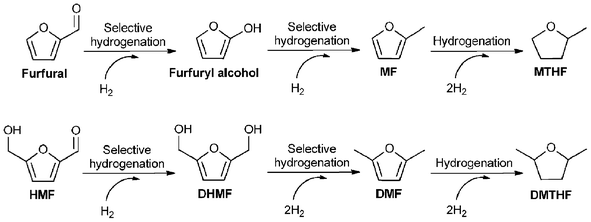 | ||
| Scheme 5 Reaction pathways for the hydrogenation of furfural and HMF. | ||
Cu-based catalysts are generally used for the hydrogenation of furfural into MF through furfuryl alcohol as an intermediate. In the earlier studies, Raney-Cu, Cu-chromite, Cu–Zn–Al–Ca–Na, Cu/Al2O3, Cu–Fe/SiO2 and Cu-chromite/C showed 87–98% MF yields at 200–300 °C with 0.15–0.3 h−1 LHSV (liquid hourly space velocity), 0.1 MPa and 5–25 H2/furfural molar ratio.182–185 However, these catalysts were reported to deactivate rapidly, which is most likely caused by thermal polymerization and coking of furfuryl alcohol on the surface of catalysts under high temperature.50 Subsequently, a novel process involving the coupling of the dehydrogenation of 1,4-butanediol (BDO) or cyclohexanol (CHL) and the hydrogenation of furfural using Cu–Zn or Cu–Zn–Al as catalysts has been reported, respectively.186,187 The coupled process could be conducted at a lower temperature (typically lower by 10–20 °C) than conventional processes while also leading to higher MF yields of 96.5 and 92.8%, respectively, which was thought to be ascribed to that the rich activated hydrogen species on the surface of catalysts from BDO or CHL dehydrogenation may promote furfural hydrogenation reaction.186,187 More recently, a few papers reported the hydrogenation of furfural into MF under milder temperatures. Sun et al.188 claimed that a yield of MF of 100% from furfural at 18 °C for 57 min with 0.1 MPa H2 using Pd complex/SiO2 as catalyst. At the same time, a variety of solvents was screened, and the best results were obtained from small, polar alcohols such as methanol and ethanol. Lange et al.50 studied the synthesis of MF from furfural, 50% yield was observed in ethanol at 30 °C for 1 h with 0.25 MPa H2 by the use of Pd/C or Pd/SiO2 in the presence of a small amount of HCl that could depress undesired side reactions. More interestingly, Li et al.189 investigated the electrocatalytic hydrogenation of furfural into MF using the cathode metal as catalysts. Among the tested anode and cathode metals, a combination of Ni anode and Ni cathode exhibited the best result. In addition, the formation of MF was favored at pH 1.0.
In 2007, Román-Leshkov et al.42 reported the synthesis of DMF from HMF using Cu–Ru/C as catalyst, a yield of 71% was achieved in 1-butanol at 220 °C for 10 h with 6.8 bar H2. Two years later, in the same reaction conditions, Binder et al.7 studied the hydrogenation of crude HMF obtained from corn stover in the presence of Cu–Ru/C, leading to 49% DMF yield. In the same year, Luijkx et al.189 obtained 35.7% DMF yield from HMF in 1-propanol at 60 °C for 150 min with 0.1 MPa H2 by the use of Pd/C. Recently, the hydrogenation of HMF was investigated in [EMIM]Cl using ACN as co-solvent, a yield of DMF with only 15% was observed using Pd/C as catalyst at 120 °C for 60 min with 68 bar H2.179 The lower yield of DMF in this work was thought to be attributed to the lower temperature and the shorter time as well as the low solubility of H2 in ionic liquid. In addition, this work also found that the source of HMF had very little effect on the conversion of HMF or the distribution of products.179 More importantly, a breakthrough in the synthesis of DMF was reported by Thananatthanachon et al.190 An excellent yield of DMF with up to 95% from HMF was reached in the presence of HCOOH, H2SO4, THF and Pd/C under reflux for 15 h. When the reaction was started from fructose, a DMF yield of 51% was also achieved. In this one-pot synthesis of DMF, HCOOH could be used not only as an acid catalyst for the dehydration of fructose into HMF and as a hydrogen source for the hydrogenation of HMF into DHMF but also as a reagent for the deoxygenation of furanylmethanols.190 Furthermore, DMF can also be produced from the selective hydrogenation of CMF.45
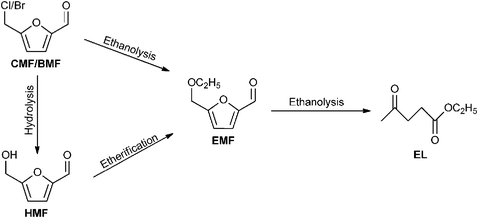 | ||
| Scheme 6 Reaction pathways for the conversion of BMF, CMF and HMF into EMF. | ||
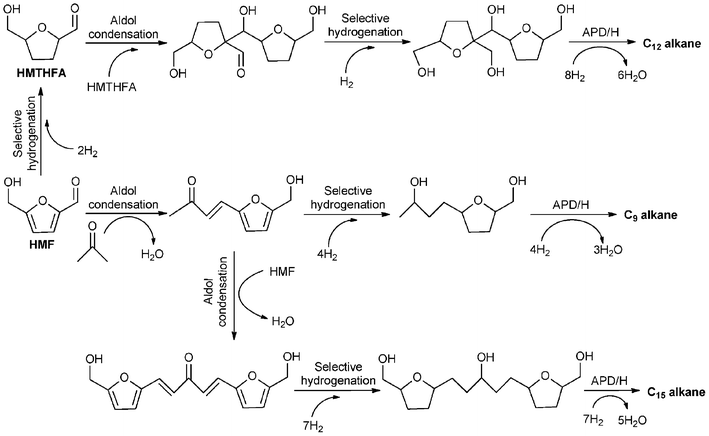
4.2. Chemicals
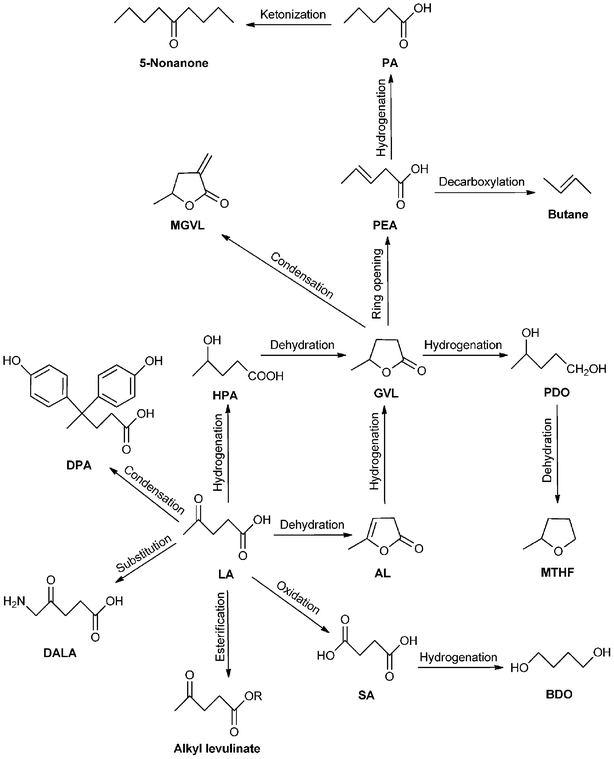 | ||
| Scheme 8 Reaction pathways for the conversion of LA into fuels and chemicals. | ||
 | ||
| Scheme 9 Reaction pathways for the oxidation of HMF into DFF and FDCA. Adapted from ref. 249. | ||
5. Conclusion and perspectives
Over the past decades, the catalytic conversion of biomass-derived carbohydrates into fuels and chemicals via furanic aldehydes has attracted considerable concern in both scientific and industrial communities. Although recent advancement has shown some exciting results, the large-scale production of furanic aldehydes-based fuels and chemicals has not been accomplished until now. In order to speed up this process, some specific points should be addressed in the future studies as follows: (1) In-depth investigation of the reaction mechanisms and kinetics for the dehydration of biomass-derived carbohydrates by the joint use of nuclear magnetic resonance, density functional theory, quantum mechanics, molecular dynamics and energetics; (2) strategic design and preparation of the more cheaper, less corrosive, lower viscous and biodegradable ionic liquids as well as the more green, active and stable solid catalysts; (3) an effective combination of solid catalysts, ionic liquids and microwave irradiation for the faster and more convenient production of furanic aldehydes; (4) A one-pot synthesis of furanic aldehydes-based fuels and chemicals directly from biomass-derived carbohydrates or lignocellulosic biomass, which can reduce the steps of the separation of furanic aldehydes and the corresponding costs; (5) the establishment of high-efficiency and energy-efficient separation and purification technologies such as membrane separation and supercritical carbon dioxide extraction technologies for desired products; (6) a complete techno-economic evaluation of the overall complexity processes for the production of furanic aldehydes and related fuels and chemicals; (7) the profound assessment of furanic aldehydes and related fuels and chemicals in order to ascertain that these products are not harmful to humans and environment; (8) the optimization of various reaction conditions using response surface analytical method or neural network algorithm and the development of continuous production technologies. All in all, we must redouble our efforts in looking for cost-effective, environmentally acceptable and workable approaches for the industrial production of fuels and chemicals from biomass-derived carbohydrates via furanic aldehydes. Despite facing many difficulties and challenges, we still believe that the future is remarkably bright.Acknowledgements
The authors gratefully acknowledge financial support of the National Basic Research Program of China (973 project, 2010CB732201), the National Natural Science Foundation of China (21106121), the Provincial R&D Program from Economic and Trade Committee of Fujian Province of China (1270-K42004), the Fundamental Research Funds for the Central Universities (2010121077) and the Fundamental Research Funds for Xiamen University (201212G006).References
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC.
- J. A. Geboers, S. V. D. Vyver, R. Ooms, B. O. D. Beeck, P. A. Jacobs and B. F. Sels, Catal. Sci. Technol., 2011, 1, 714–726 RSC.
- H. Kobayashi, H. Ohta and A. Fukuoka, Catal. Sci. Technol., 2012, 2, 869–883 RSC.
- A. A. Rosatella, S. P. Simeonov, R. F. M. Fradea and C. A. M. Afonso, Green Chem., 2011, 13, 754–793 RSC.
- X. L. Tong, Y. Ma and Y. D. Li, Appl. Catal., A, 2010, 385, 1–13 CrossRef CAS.
- C. H. Zhou, X. Xia, C. X. Lin, D. S. Tong and J. Beltramini, Chem. Soc. Rev., 2011, 40, 5588–5617 RSC.
- J. B. Binder and R. T. Raines, J. Am. Chem. Soc., 2009, 131, 1979–1985 CrossRef CAS.
- Q. Cao, X. C. Guo, J. Guan, X. D. Mu and D. K. Zhang, Appl. Catal., A, 2011, 403, 98–103 Search PubMed.
- C. Perego and A. Bosetti, Microporous Mesoporous Mater., 2011, 144, 28–39 CrossRef CAS.
- X. H. Qi, H. X. Guo and L. Y. Li, Ind. Eng. Chem. Res., 2011, 50, 7985–7989 CrossRef CAS.
- P. Wang, H. B. Yu, S. H. Zhan and S. Q. Wang, Bioresour. Technol., 2011, 102, 4179–4183 CrossRef CAS.
- J. C. Serrano-Ruiz and J. A. Dumesic, Energy Environ. Sci., 2011, 4, 83–99 RSC.
- S. Lima, M. M. Antunes, M. Pillinger and A. A. Valente, ChemCatChem, 2011, 3, 1686–1706 Search PubMed.
- T. Stahlberg, W. J. Fu and J. M. Woodley, ChemSusChem, 2011, 4, 451–458 CrossRef CAS.
- M. E. Zakrzewska, E. Bogel-Lukasik and R. Bogel-Lukasik, Chem. Rev., 2011, 111, 397–417 CrossRef CAS.
- X. H. Qi, M. Watanabe, T. M. Aida and R. L. Smith, Bioresour. Technol., 2012, 109, 224–228 Search PubMed.
- O. O. James, S. Maity, L. A. Usman, K. O. Ajanaku and O. O. Ajani, Energy Environ. Sci., 2010, 3, 1833–1850 RSC.
- R. Karinen, K. Vilonen and M. Niemela, ChemSusChem, 2011, 4, 1002–1016 CrossRef CAS.
- G. Liu, J. M. Wu, I. Y. Zhang, Z. N. Chen, Y. W. Li and X. Xu, J. Phys. Chem. A, 2011, 115, 13628–13641 Search PubMed.
- J. C. Serrano-Ruiz, R. Luque and A. Sepulveda-Escribano, Chem. Soc. Rev., 2011, 40, 5266–5281 RSC.
- E. Taarning, C. M. Osmundsen, X. B. Yang, B. Voss and S. I. Andersen, Energy Environ. Sci., 2011, 4, 793–804 RSC.
- J. B. Binder, J. J Blank, A. V. Cefali and R. T. Raines, ChemSusChem, 2010, 3, 1268–1272 CrossRef CAS.
- E. Lam, J. H. Chong, E. Majid, Y. L. Liu, S. Hrapovic, A. C. W. Leung and J. H. T. Luong, Carbon, 2012, 50, 1033–1043 CrossRef CAS.
- X. J. Shi, Y. L. Wu, P. P. Li, H. F. Yi, M. D. Yang and G. H. Wang, Carbohydr. Res., 2011, 346, 480–487 Search PubMed.
- J. H. Zhang, J. P. Zhuang, L. Lin, S. J. Liu and Z. Zhang, Biomass Bioenergy, 2012, 39, 73–77 CrossRef CAS.
- S. Sitthisa and D. E. Resasco, Catal. Lett., 2011, 141, 784–791 CrossRef CAS.
- R. M. West, Z. Y. Liu, M. Peter and C. A. Gärtner, J. Mol. Catal. A: Chem., 2008, 296, 18–27 CrossRef CAS.
- X. H. Zhang, T. J. Wang, L. L. Ma and C. Z. Wu, Fuel, 2010, 89, 2697–2702 Search PubMed.
- M. G. Kim, G. Boyd and R. Strickland, Holzforschung, 1994, 48, 262–267 Search PubMed.
- S. Shi, H. Guo and G. Yin, Catal. Commun., 2011, 12, 731–733 CrossRef CAS.
- A. S. Mamman, J. M. Lee, Y. C. Kim, I. T. Hwang and N. J. Park, Biofuels, Bioprod. Biorefin., 2008, 2, 438–454 Search PubMed.
- T. S. Hansen, J. Mielby and A. Riisager, Green Chem., 2011, 13, 109–114 RSC.
- S. Q. Hu, Z. F. Zhang, J. L. Song, Y. X. Zhou and B. X. Han, Green Chem., 2009, 11, 1746–1749 RSC.
- X. H. Qi, M. Watanabe, T. M. Aida and R. L. Smith, ChemSusChem, 2010, 3, 1071–1077 CrossRef CAS.
- Y. Román-Leshkov, J. N. Chheda and J. A. Dumesic, Science, 2006, 312, 1933–1937 CrossRef CAS.
- K. D. O. Vigier, A. Benguerba, J. Barrault and F. Jerome, Green Chem., 2012, 14, 285–289 RSC.
- G. Yong, Y. G. Zhang and J. Y. Ying, Angew. Chem., 2008, 120, 9485–9488 CrossRef.
- H. B. Zhao, J. E. Holladay, H. Brown and Z. C. Zhang, Science, 2007, 316, 1597–1600 CrossRef CAS.
- Y. Román-Leshkov, C. J. Barrett and Z. Y. Liu, Nature, 2007, 447, 982–986 CrossRef CAS.
- W. R. Yang and A. Sen, ChemSusChem, 2010, 3, 597–603 CrossRef CAS.
- X. Xiang, L. He, Y. Yang, B. Guo, D. M. Tong and C. W. Hu, Catal. Lett., 2011, 141, 735–741 CrossRef CAS.
- N. K. Gupta, S. Nishimura, A. Takagaki and K. Ebitani, Green Chem., 2011, 13, 824–827 RSC.
- L. C. Peng, L. Lin, J. H. Zhang, J. P. Zhuang, B. X. Zhang and Y. Gong, Molecules, 2010, 15, 5258–5272 Search PubMed.
- G. W. Huber, J. N. Chheda, C. J. Barrett and J. A. Dumesic, Science, 2005, 308, 1446–1450 CrossRef CAS.
- M. Mascal and E. B. Nikitin, Angew. Chem., Int. Ed., 2008, 47, 7924–7926 CrossRef CAS.
- M. Mascal and E. B. Nikitin, ChemSusChem, 2009, 2, 423–426 CrossRef CAS.
- M. Mascal and E. B. Nikitin, ChemSusChem, 2009, 2, 859–861 CrossRef CAS.
- M. Mascal and E. B. Nikitin, Green Chem., 2010, 12, 370–373 RSC.
- N. Kumari, J. K. Olesen and C. M. Pedersen, Eur. J. Org. Chem., 2011, 1266–1270 CrossRef CAS.
- J. P. Lange, E. van der Heide and J. van Buijtenen, ChemSusChem, 2012, 5, 150–166 CrossRef CAS.
- M. J. Antal, T. Leesomboon, W. S. Mok and G. N. Richards, Carbohydr. Res., 1991, 217, 71–85 CrossRef.
- J. N. Chheda, Y. Román-Leshkov and J. A. Dumesic, Green Chem., 2007, 9, 342–350 RSC.
- D. Montané, J. Salvadó and C. Torras, Biomass Bioenergy, 2002, 22, 295–304 CrossRef CAS.
- R. Weingarten, J. Cho, W. C. Conner and G. W. Huber, Green Chem., 2010, 12, 1423–1429 RSC.
- W. Riansa-Ngawong and P. Prasertsan, Carbohydr. Res., 2011, 346, 103–110 Search PubMed.
- O. Yemis and G. Mazza, Bioresour. Technol., 2011, 102, 7371–7378 CrossRef CAS.
- O. Yemis and G. Mazza, Bioresour. Technol., 2012, 109, 215–223 Search PubMed.
- R. O'Neill, M. N. Ahmad, L. Vanoye and F. Aiouache, Ind. Eng. Chem. Res., 2009, 48, 4300–4306 CrossRef CAS.
- P. L. Dhepe and R. A. Sahu, Green Chem., 2010, 12, 2153–2156 RSC.
- V. Choudhary, A. B. Pinar, S. I. Sandler, D. G. Vlachos and R. F. Lobo, ACS Catal., 2011, 1, 1724–1728 Search PubMed.
- G. H. Jeong, E. G. Kim, S. B. Kim, E. D. Park and S. W. Kim, Microporous Mesoporous Mater., 2011, 144, 134–139 CrossRef CAS.
- A. Takagaki, M. Ohara, S. Nishimura and K. Ebitani, Chem. Lett., 2010, 39, 838–840 CrossRef CAS.
- E. Lam, J. H. Chong, E. Majid, Y. L. Liu, S. Hrapovic, A. C. W. Leung and J. H. T. Luong, ChemSusChem, 2011, 4, 535–541 CrossRef CAS.
- C. Moreau, R. Durand, D. Peyron, J. Duhamet and P. Rivalier, Ind. Crops Prod., 1998, 7, 95–99 CrossRef CAS.
- I. Agirrezabal-Telleria, A. Larreategui, J. Requies, M. B. Güemez and P. L. Arias, Bioresour. Technol., 2011, 102, 7478–85 CrossRef CAS.
- A. S. Dias, M. Pillinger and A. A. Valente, J. Catal., 2005, 229, 414–423 CrossRef CAS.
- A. S. Dias, S. Lima, D. Carriazo, V. Rives, M. Pillinger and A. A. Valente, J. Catal., 2006, 244, 230–237 CrossRef CAS.
- A. S. Dias, M. Pillinger and A. A. Valente, Microporous Mesoporous Mater., 2006, 94, 214–225 CrossRef CAS.
- A. S. Dias, S. Lima, V. Rives, M. Pillinger and A. A. Valente, Carbohydr. Res., 2006, 341, 2946–2953 CrossRef CAS.
- S. B. Kim, S. J. You, Y. T. Kim, S. M. Lee, H. Lee, K. Park and E. D. Park, Korean J. Chem. Eng., 2011, 28, 710–716 Search PubMed.
- S. Lima, M. Pillinger and A. A. Valente, Catal. Commun., 2008, 9, 2144–2148 CrossRef CAS.
- S. Lima, A. Fernandes, M. M. Antunes, M. Pillinger, F. Ribeiro and A. A. Valente, Catal. Lett., 2010, 135, 41–47 CrossRef CAS.
- J. Lessard, J. F. Morin, J. F. Wehrung, D. Magnin and E. Chornet, Top. Catal., 2010, 53, 1231–1234 CrossRef CAS.
- I. Sádabaa, S. Lima and A. A. Valente, Carbohydr. Res., 2011, 346, 2785–2791 Search PubMed.
- X. J. Shi, Y. L. Wu, P. P. Li, H. F. Yi, M. D. Yang and G. H. Wang, Energies, 2011, 4, 669–684 Search PubMed.
- T. Suzuki, T. Yokoi, R. Otomo and J. N. Kondo, Appl. Catal., A, 2011, 408, 117–124 CrossRef CAS.
- W. Daengprasert, P. Boonnoun, N. Laosiripojana, M. Goto and A. Shotipruk, Ind. Eng. Chem. Res., 2011, 50, 7903–7910 Search PubMed.
- M. M. Antunesa, S. Lima, A. Fernandes, M. Pillinger, M. F. Ribeiro and A. A. Valente, Appl. Catal., A, 2012, 417–418, 243–252 Search PubMed.
- S. Lima, P. Neves, M. M. Antunes, M. Pillinger, N. Ignatyev and A. A. Valente, Appl. Catal., A, 2009, 363, 93–99 CrossRef CAS.
- F. R. Tao, H. L. Song and L. J. Chou, Can. J. Chem., 2011, 89, 83–87 CrossRef CAS.
- Z. H. Zhang and Z. B. Zhao, Bioresour. Technol., 2010, 101, 1111–1114 CrossRef CAS.
- M. R. Nimlos, X. H. Qian, M. Davis, M. E. Himmel and D. K. Johnson, J. Phys. Chem. A, 2006, 110, 11824–11838 CrossRef.
- T. Ahmad, L. Kenne, K. Olsson and O. Theander, Carbohydr. Res., 1995, 276, 309–320 CrossRef CAS.
- M. S. Feather, Tetrahedron Lett., 1970, 11, 4143–4145 Search PubMed.
- M. S. Feather, D. W. Harris and S. B. Nichols, J. Org. Chem., 1972, 37, 1606–1608 CrossRef CAS.
- G. Marcotullio and W. De Jong, Green Chem., 2010, 12, 1739–1746 RSC.
- G. Marcotullio and W. De Jong, Carbohydr. Res., 2011, 346, 1291–1293 CrossRef CAS.
- Q. Cao, X. C. Guo, S. X. Yao, J. Guan, X. Y. Wang, X. D. Mu and D. K. Zhang, Carbohydr. Res., 2011, 346, 956–959 CrossRef CAS.
- B. R. Caes and R. T. Raines, ChemSusChem, 2011, 4, 353–356 CrossRef CAS.
- C. Z. Li, Z. B. Zhao, A. Q. Wang, M. Y. Zheng and T. Zhang, Carbohydr. Res., 2010, 345, 1846–1850 CrossRef CAS.
- Y. H. Li, X. Y. Lu, L. Yuan and X. Liu, Biomass Bioenergy, 2009, 33, 1182–1187 CrossRef CAS.
- L. K. Lai and Y. G. Zhang, ChemSusChem, 2010, 3, 1257–1259 CrossRef CAS.
- L. K. Lai and Y. G. Zhang, ChemSusChem, 2011, 4, 1745–1748 CrossRef CAS.
- Y. Román-Leshkov and J. A. Dumesic, Top. Catal., 2009, 52, 297–303 CrossRef CAS.
- M. Watanabe, Y. Aizawa, T. Iida and T. M. Aida, Carbohydr. Res., 2005, 340, 1925–1930 CrossRef CAS.
- Y. B. Yi, J. W. Lee, S. S. Hong, Y. H. Choi and C. H. Chung, J. Ind. Eng. Chem., 2011, 17, 6–9 Search PubMed.
- X. H. Qi, M. Watanabe, T. M. Aida and R. L. Smith, Green Chem., 2008, 10, 799–805 RSC.
- X. H. Qi, M. Watanabe, T. M. Aida and R. L. Smith, Ind. Eng. Chem. Res., 2008, 47, 9234–9239 CrossRef CAS.
- K. I. Shimizu, R. Uozumi and A. Satsuma, Catal. Commun., 2009, 10, 1849–1853 CrossRef CAS.
- X. H. Qi, M. Watanabe, T. M. Aida and R. L. Smith, Green Chem., 2009, 11, 1327–1331 RSC.
- H. Zhu, Q. Cao, C. H. Li and X. D. Mu, Carbohydr. Res., 2011, 346, 2016–2018 Search PubMed.
- C. Moreau, R. Durand, S. Razigade, J. Duhamet, P. Faugeras, P. Rivalier, P. Ros and G. Avignon, Appl. Catal., A, 1996, 145, 211–224 CrossRef CAS.
- V. V. Ordomsky, J. van der Schaaf, J. C. Schouten and T. A. Nijhuis, J. Catal., 2012, 287, 68–75 CrossRef CAS.
- X. C. Guo, Q. Cao, Y. J. Jiang, J. Guan, X. Y. Wang and X. D. Mu, Carbohydr. Res., 2012, 351, 35–41 Search PubMed.
- X. H. Qi, M. Watanabe, T. M. Aida and R. L. Smith, Catal. Commun., 2008, 9, 2244–2249 CrossRef CAS.
- X. H. Qi, M. Watanabe, T. M. Aida and R. L. Smith, Catal. Commun., 2009, 10, 1771–1775 CrossRef CAS.
- D. Stosic, S. Bennici, V. RakIc and A. Auroux, Catal. Today, 2011 DOI:10.1016/j.cattod.2011.10.040.
- S. Dutta, S. De, A. K. Patra, M. Sasidharan, A. Bhaumik and B. Saha, Appl. Catal., A, 2011, 409–410, 133–139 CrossRef CAS.
- F. L. Yang, Q. S. Liu, M. Yue, X. F. Bai and Y. G. Du, Chem. Commun., 2011, 47, 4469–4471 RSC.
- D. Ray, N. Mittal and W. J. Chung, Carbohydr. Res., 2011, 346, 2145–2148 Search PubMed.
- C. Y. Fan, H. Y. Guan, H. Zhang, J. H. Wang, S. T. Wang and X. H. Wang, Biomass Bioenergy, 2011, 35, 2659–2665 CrossRef CAS.
- Q. Zhao, L. Wang, S. Zhao, X. H. Wang and S. T. Wang, Fuel, 2011, 90, 2289–2293 Search PubMed.
- Y. M. Zhang, V. Degirmenci, C. Li and E. J. M. Hensen, ChemSusChem, 2011, 4, 59–64 CrossRef CAS.
- Y. S. Qu, C. P. Huang, J. Zhang and B. H. Chen, Bioresour. Technol., 2012, 106, 170–172 CrossRef CAS.
- F. Ilgen, D. Ott, D. Kralisch, C. Reil, A. Palmberger and B. Konig, Green Chem., 2009, 11, 1948–1954 RSC.
- Z. S. Yuan, C. B. Xu, S. N. Cheng and M. Leitch, Carbohydr. Res., 2011, 346, 2019–2023 Search PubMed.
- S. De, S. Dutta and S. B. Saha, Green Chem., 2011, 13, 2859–2868 RSC.
- X. L. Tong, M. R. Li, N. Yan, Y. Ma, P. J. Dyson and Y. D. Li, Catal. Today, 2011, 175, 524–527 Search PubMed.
- Z. J. Wei, Y. Li, D. Thushara, Y. X. Liu and Q. L. Ren, J. Taiwan Inst. Chem. Eng., 2011, 42, 363–370 Search PubMed.
- Z. J. Wei, Y. X. Liu, D. Thushara and Q. L. Ren, Green Chem., 2012, 14, 1220–1226 RSC.
- Z. H. Zhang, Q. Wang, H. B. Xie, W. J. Liu and Z. B. Zhao, ChemSusChem, 2011, 4, 131–138 CrossRef CAS.
- F. F. Wang, A. W. Shi, X. X. Qin, C. L. Liu and W. S. Dong, Carbohydr. Res., 2011, 346, 982–985 Search PubMed.
- J. T. Liu, Y. Tang, K. G. Wu, C. F. Bi and Q. Cui, Carbohydr. Res., 2012, 350, 20–24 Search PubMed.
- J. Y. G. Chan and Y. Zhang, ChemSusChem, 2009, 2, 731–734 CrossRef.
- C. Moreau, A. Finiels and L. Vanoye, J. Mol. Catal. A: Chem., 2006, 253, 165–169 CrossRef CAS.
- S. Q. Hu, Z. F. Zhang, Y. X. Zhou, B. X. Han, H. L. Fan, W. J. Li, J. L. Song and Y. Xie, Green Chem., 2008, 10, 1280–1283 RSC.
- S. Q. Hu, Z. F. Zhang, Y. X. Zhou, J. L. Song, H. L. Fan and B. X. Han, Green Chem., 2009, 11, 873–877 RSC.
- X. L. Tong and Y. D. Li, ChemSusChem, 2010, 3, 350–355 CAS.
- A. H. Jadhav and H. Kim, Catal. Commun., 2012, 21, 96–103 CrossRef CAS.
- Q. X. Bao, K. Qiao, D. Tomida and C. Yokoyama, Catal. Commun., 2008, 9, 1383–1388 CrossRef CAS.
- K. B. Sidhpuria, A. L. Daniel-da-Silva, T. Trindade and J. A. P. Coutinho, Green Chem., 2011, 13, 340–349 RSC.
- T. Caruso and E. Vasca, Electrochem. Commun., 2010, 12, 1149–1153 Search PubMed.
- C. Z. Li, Z. B. Zhao, H. L. Cai, A. Q. Wang and T. Zhang, Biomass Bioenergy, 2011, 35, 2013–2017 CrossRef.
- J. Ryu, J. W. Choi, D. J. Suh, D. J. Ahn and Y. W. Suh, Catal. Commun., 2012, 24, 11–15 CrossRef CAS.
- H. P. Yan, Y. Yang, D. M. Tong, X. Xiang and C. W. Hu, Catal. Commun., 2009, 10, 1558–1563 CrossRef CAS.
- X. L. Tong, Y. Ma and Y. D. Li, Carbohydr. Res., 2010, 345, 1698–1701 CrossRef CAS.
- Y. M. Zhang, E. A. Pidko and E. J. M. Hensen, Chem.–Eur. J., 2011, 17, 5281–5288 CrossRef CAS.
- T. M. Chen and L. Lin, Chin. J. Chem., 2010, 28, 1773–1776 Search PubMed.
- L. Hu, Y. Sun and L. Lin, Ind. Eng. Chem. Res., 2012, 51, 1099–1104 Search PubMed.
- H. Jadhav, E. Taarning, C. M. Pedersen and M. Bols, Tetrahedron Lett., 2012, 53, 983–985 Search PubMed.
- Y. Yang and C. W. Hu, Green Chem., 2012, 14, 509–513 RSC.
- M. Yasuda, Y. Nakamura, J. Matsumoto, H. Yokoi and T. Shiragami, Bull. Chem. Soc. Jpn., 2011, 84, 416–418 Search PubMed.
- K. Beckerle and K. Okuda, J. Mol. Catal. A: Chem., 2012, 356, 158–164 Search PubMed.
- T. Stahlberg, M. G. Sørensen and A. Riisager, Green Chem., 2010, 12, 321–325 RSC.
- K. Nakajima, Y. Baba, R. Noma, M. Kitano, J. N. Kondo, S. Hayas and M. Hara, J. Am. Chem. Soc., 2011, 133, 4224–4227 CrossRef CAS.
- F. L. Yang, Q. S. Liu, X. F. Bai and Y. G. Du, Bioresour. Technol., 2011, 102, 3424–3429 CrossRef CAS.
- M. Kitano, K. Nakajima, J. N. Kondo, S. Hayashi and M. Hara, J. Am. Chem. Soc., 2010, 132, 6622–6623 CrossRef.
- V. Degirmenci, E. A. Pidko, P. C. M. M. Magusin and E. J. M. Hensen, ChemCatChem, 2011, 3, 969–972 Search PubMed.
- F. Guo, Z. Fang and T. J. Zhou, Bioresour. Technol., 2012, 112, 313–318 Search PubMed.
- M. Ohara, A. Takagaki, S. Nishimura and K. Ebitani, Appl. Catal., A, 2010, 383, 149–155 CrossRef CAS.
- A. Takagaki, M. Ohara, S. Nishimura and K. Ebitani, Chem. Commun., 2009, 6276–6278 RSC.
- J. Tuteja, S. Nishimura and K. Ebitani, Bull. Chem. Soc. Jpn., 2012, 85, 275–281 CrossRef CAS.
- E. Nikolla, Y. Román-Leshkov, M. Moliner and M. E. Davis, ACS Catal., 2011, 1, 408–410 Search PubMed.
- P. Azadi, R. Carrasquillo-Flores, Y. J. Pagán-Torres, E. I. Gürbüz, R. Farnood and J. A. Dumesic, Green Chem., 2012, 14, 1573–1576 RSC.
- R. L. Huang, W. Qi, R. X. Su and Z. M. He, Chem. Commun., 2010, 46, 1115–1117 RSC.
- T. Stahlberg, S. Rodriguez-Rodriguez, P. Fristrup and A. Riisager, Chem.–Eur. J., 2011, 17, 1456–1464 CrossRef CAS.
- E. A. Khokhlova, V. V. Kachala and V. P. Ananikov, ChemSusChem, 2012, 5, 783–789 Search PubMed.
- J. A. Chun, J. W. Lee, Y. B. Yi, S. S. Hong and C. H. Chung, Korean J. Chem. Eng., 2010, 27, 930–935 Search PubMed.
- C. Wang, L. T. Fu, X. L. Tong, Q. W. Yang and W. Q. Zhang, Carbohydr. Res., 2012, 347, 182–185 Search PubMed.
- Y. T. Zhang, H. B. Du, X. H. Qian and E. Y. X. Chen, Energy Fuels, 2010, 24, 2410–2417 CrossRef CAS.
- Y. Su, H. M. Brown, X. W. Huang, X. D. Zhou, J. E. Amonette and Z. C. Zhang, Appl. Catal., A, 2009, 361, 117–122 CrossRef CAS.
- B. Kim, J. Jeong, D. Lee, S. Y. Kim, H. J. Yoon, Y. S. Lee and J. K. Cho, Green Chem., 2011, 13, 1503–1506 RSC.
- S. Dutta, S. De, M. Imteyaz Alam, M. M. Abu-Omar and B. Saha, J. Catal., 2012, 288, 8–15 CrossRef CAS.
- S. Zhao, M. X. Cheng, J. Z. Li, J. Tian and X. H. Wang, Chem. Commun., 2011, 47, 2176–2178 RSC.
- A. S. Amarasekara, L. T. D. Williams and C. C. Ebede, Carbohydr. Res., 2008, 343, 3021–3024 CrossRef CAS.
- M. J. Antal, W. S. L. Mok and G. N. Richards, Carbohydr. Res., 1990, 199, 91–109 CrossRef CAS.
- R. S. Assary, P. C. Redfern, J. Greeley and L. A. Curtiss, J. Phys. Chem. B, 2011, 115, 4341–4349 CrossRef CAS.
- J. Guan, Q. Cao, X. C. Guo and X. D. Mu, Comput. Theor. Chem., 2011, 963, 453–462 Search PubMed.
- X. H. Qian, Top. Catal., 2012, 55, 218–226 Search PubMed.
- J. B. Binder, A. V. Cefali, J. J. Blankc and R. T. Raines, Energy Environ. Sci., 2010, 3, 765–771 RSC.
- M. Moliner, Y. Román-Leshkov and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6164–6168 CrossRef CAS.
- E. A. Pidko, V. Degirmenci, R. A. Van Santen and E. J. M. Hensen, Angew. Chem., Int. Ed., 2010, 49, 2530–2534 CrossRef CAS.
- Y. Román-Leshkov, M. Moliner, J. A. Labinger and M. E. Davis, Angew. Chem., Int. Ed., 2010, 49, 8954–8957 CrossRef CAS.
- M. Brasholz, K. V. Kanel, C. H. Hornung, S. Saubern and T. J. Sana, Green Chem., 2011, 13, 1114–1117 RSC.
- S. Mallakpour and Z. Rafieec, Prog. Polym. Sci., 2011, 36, 1754–1765 Search PubMed.
- C. Z. Li, Z. H. Zhang and Z. B. K. Zhao, Tetrahedron Lett., 2009, 50, 5403–5405 CrossRef CAS.
- L. Hu, Y. Sun and L. Lin, Prog. Chem., 2012, 24, 483–491 Search PubMed.
- M. E. Zakrzewska, E. Bogel-Lukasik and R. Bogel-Lukasik, Energy Fuels, 2010, 24, 737–745 CrossRef CAS.
- M. Chidambaram and A. T. Bell, Green Chem., 2010, 12, 1253–1262 RSC.
- S. H. Mushrif, S. Caratzoulas and D. G. Vlachos, Phys. Chem. Chem. Phys., 2012, 14, 2637–2644 RSC.
- N. Guo, S. Caratzoulas, D. J. Doren, S. I. Sandler and D. G. Vlachos, Energy Environ. Sci., 2012, 5, 6703–6716 RSC.
- L. W. Burnett, I. B. Johns, R. F. Holdren and R. M. Hixon, Ind. Eng. Chem., 1948, 40, 502–505 CrossRef CAS.
- J. G. M. Bremner and R. K. F. Keeys, J. Chem. Soc., 1947, 1068–1080 Search PubMed.
- J. Lessard, J. F. Morin, J. F. Wehrung, D. Magnin and E. Chornet, Top. Catal., 2010, 53, 1231–1234 CrossRef CAS.
- H. Y. Zheng, Y. L. Zhu, B. T. Teng, Z. Q. Bai, C. H. Zhang, H. W. Xiang and Y. W. Li, J. Mol. Catal. A: Chem., 2006, 246, 18–23 CrossRef CAS.
- Y. L. Zhu, H. W. Xiang, Y. W. Li, H. J. Jiao, G. S. Wu, B. Zhong and G. Q. Guo, New J. Chem., 2003, 27, 208–210 RSC.
- H. Y. Zhang, Y. L. Zhu, Z. Q. Bai, L. Huang, H. W. Xiang and Y. W. Li, Green Chem., 2006, 8, 107–109 RSC.
- Q. Sun, S. F. Liu, X. H. Yao, Y. C. Su and Z. Q. Zhang, Chin. J. Syn. Chem., 1996, 4, 146–150 Search PubMed.
- G. C. A. Luijkx, N. P. M. Huck, F. Van Rantwijk, L. Maat and H. Van Bekkum, Heterocycles, 2009, 77, 1037–1044 CrossRef CAS.
- T. Thananatthanachon and T. B. Rauchfuss, Angew. Chem., Int. Ed., 2010, 49, 6616–6618 CrossRef CAS.
- M. Balakrishnan, E. R. Sacia and A. T. Bell, Green Chem., 2012, 14, 1626–1634 RSC.
- P. Lanzafame, D. M. Temi, S. Perathoner, G. Centi, A. Macario, A. Aloise and G. Giordano, Catal. Today, 2011, 175, 435–441 CrossRef CAS.
- C. J. Barrett, J. N. Chheda, G. W. Huber and J. A. Dumesic, Appl. Catal., B, 2006, 66, 111–118 CrossRef CAS.
- N. Fakhfakh, P. Cognet, M. Cabassud and Y. Lucchese, Chem. Eng. Process., 2008, 47, 349–362 CrossRef CAS.
- L. Faba, E. Díaz and S. Ordóñez, Appl. Catal., B, 2012, 113–114, 201–211 Search PubMed.
- I. Sádaba, M. Ojeda, R. Mariscal, J. L .G. Fierro and M. L. Granados, Appl. Catal., B, 2011, 101, 638–648 Search PubMed.
- W. Q. Shen, G. A. Tompsetta, K. D. Hammonda, R. Xing, F. Dogan, C. P. Grey, W. C. Conner, S. M. Auerbacha and G. W. Huber, Appl. Catal., A, 2011, 392, 57–68 Search PubMed.
- M. Chatterjee, K. Matsushima, Y. Ikushima, M. Sato, T. Yokoyama, H. Kawanami and T. Suzuki, Green Chem., 2010, 12, 779–782 RSC.
- R. Xing, A. V. Subrahmanyam, H. Olcay, W. Qi, G. P. Walsum, H. Pends and G. W. Huber, Green Chem., 2010, 12, 1933–1946 RSC.
- J. N. Chheda and J. A. Dumesic, Catal. Today, 2007, 123, 59–70 CrossRef CAS.
- J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164–7183 CrossRef CAS.
- W. Dedsuksophon, F. K. aungnawakij, V. Champreda and N. Laosiripojana, Bioresour. Technol., 2011, 102, 2040–2046 CrossRef CAS.
- W. J. Xu, X. H. Liu, J. W. Ren, P. Zhang, Y. Q. Wang, Y. L. Guo, Y. Guo and G. Z. Lu, Catal. Commun., 2010, 11, 721–726 CrossRef CAS.
- B. Girisuta, L. P. B. M. Janssen and H. J. Heeres, Green Chem., 2006, 8, 701–709 RSC.
- S. V. D. Vyver, J. Thomas, J. Geboers, S. Keyzer, M. Smet, W. Dehaen, P. A. Jacobs and B. F. Sels, Energy Environ. Sci., 2011, 4, 3601–3610 RSC.
- C. Chang, X. J. Ma and P. L. Cen, Chin. J. Chem. Eng., 2009, 17, 835–839 Search PubMed.
- B. Girisuta, L. P. B. M. Janssen and H. J. Heeres, Chem. Eng. Res. Des., 2006, 84, 339–349 CrossRef CAS.
- B. Girisuta, B. Danon, R. Manurung, L. P. B. M. Janssen and H. J. Heeres, Bioresour. Technol., 2008, 99, 8367–8375 CrossRef CAS.
- D. W. Rackemann and W. O. S. Doherty, Biofuels, Bioprod. Biorefin., 2011, 5, 198–214 Search PubMed.
- J. C. Shen and C. E. Wyman, AIChE J., 2012, 58, 236–246 CrossRef CAS.
- H. Z. Chen, B. Yu and S. Y. Jin, Bioresour. Technol., 2011, 102, 3568–3570 CrossRef CAS.
- J. Hegner, K. C. Pereira, B. DeBoef and B. L. Lucht, Tetrahedron Lett., 2010, 51, 2356–2358 CrossRef CAS.
- J. Potvin, E. Sorlien, J. Hegner, B. DeBoef and B. L. Lucht, Tetrahedron Lett., 2011, 52, 5891–5893 CrossRef CAS.
- N. Ya'aini, N. A. S. Amin and M. A. M. Yussuf, Bioresour. Technol., 2012, 116, 58–65 Search PubMed.
- S. Dharne and V. V. Bokade, J. Nat. Gas Chem., 2011, 20, 18–24 Search PubMed.
- X. Hu and C. Z. Li, Green Chem., 2011, 13, 1676–1679 RSC.
- G. Pasquale, P. Vázquez, G. Romanelli and G. Baronetti, Catal. Commun., 2012, 18, 115–120 Search PubMed.
- L. C. Peng, L. Lin, J. H. Zhang, J. B. Shi and S. J. Liu, Appl. Catal., A, 2011, 397, 259–265 CrossRef CAS.
- L. C. Peng, L. Lin, J. H. Zhang, J. B. Shi and Q. L. Yang, Appl. Energy, 2011, 88, 4590–4596 Search PubMed.
- L. C. Peng, L. Lin and H. Li, Ind. Crops Prod., 2012, 40, 136–144 Search PubMed.
- F. Rataboul and N. Essayem, Ind. Eng. Chem. Res., 2011, 50, 799–805 CrossRef CAS.
- S. Saravanamurugan and A. Riisager, Catal. Commun., 2012, 17, 71–75 CrossRef CAS.
- D. M. Alonso, S. G. Wettstein, J. Q. Bond, T. W. Root and J. A. Dumesic, ChemSusChem, 2011, 4, 1078–1081 CrossRef CAS.
- D. J. Braden, C. A. Henao, J. Heltzel, C. C. Maravelias and J. A. Dumesic, Green Chem., 2011, 13, 1755–1765 RSC.
- X. L. Du, L. He, S. Zhao, Y. M. Liu, Y. Cao, H. Y. He and K. N. Fan, Angew. Chem., Int. Ed., 2011, 50, 7815–7819 CrossRef CAS.
- A. M. R. Galletti, C. Antonetti, V. D. Luise and M. Martinelli, Green Chem., 2012, 14, 688–694 RSC.
- S. G. Wettstein, J. Q. Bond, D. M. Alonso, H. N. Pham, A. K. Datye and J. A. Dumesic, Appl. Catal., B, 2012, 117–118, 321–329 CrossRef CAS.
- Z. P. Yan, L. Lin and S. J. Liu, Energy Fuels, 2009, 23, 3853–3858 CrossRef CAS.
- J. J. Bozell, L. Moens, D. C. Elliott, Y. Wang, G. G. Neuenscwander, S. W. Fitzpatrick, R. J. Bilski and J. L. Jarnefeld, Resour., Conserv. Recycl., 2000, 28, 227–239 CrossRef.
- H. Mehdi, V. Fabos, R. Tuba, A. Bodor, L. T. Mika and I. T. Horvath, Top. Catal., 2008, 48, 49–54 CrossRef CAS.
- J. Q. Bond, D. M. Alonso, D. Wang, R. M. West and J. A. Dumesic, Science, 2010, 327, 1110–1114 CrossRef CAS.
- J. Q. Bond, D. Wang, D. M. Alonso and J. A. Dumesic, J. Catal., 2011, 281, 290–299 CrossRef CAS.
- L. E. Manzer, Appl. Catal., A, 2004, 272, 249–256 CrossRef CAS.
- A. D. Patel, J. C. Serrano-Ruiz, J. A. Dumesic and R. P. Anex, Chem. Eng. J., 2010, 160, 311–321 CrossRef CAS.
- J. C. Serrano-Ruiz, D. J. Braden, R. M. West and J. A. Dumesic, Appl. Catal., B, 2010, 100, 184–189 CrossRef CAS.
- Y. H. Guo, K. X. Li, X. D. Yu and J. H. Clark, Appl. Catal., B, 2008, 81, 182–191 CrossRef CAS.
- S. V. D. Vyver, J. Geboers, S. Helsen, F. Yu, J. Thomas, M. Smet, W. Dehaen and B. F. Sels, Chem. Commun., 2012, 48, 3497–3499 RSC.
- X. D. Yu, Y. H. Guo, K. X. Li, X. Yang, L. L. Xu, Y. N. Guo and J. L. Hu, J. Mol. Catal. A: Chem., 2008, 290, 44–53 CrossRef CAS.
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC.
- A. Gandini and M. N. Belgacem, Prog. Polym. Sci., 1997, 22, 1203–1379 CrossRef CAS.
- A. S. Amarasekara, D. Green and E. McMillan, Catal. Commun., 2008, 9, 286–288 CrossRef CAS.
- L. Cottier, G. Descotes, E. Viollet, J. Lewkowski and R. Skowronski, J. Heterocycl. Chem., 1995, 32, 927–930 CrossRef CAS.
- J. Lewkowski, ARKIVOC, 2001, 1, 17–54 Search PubMed.
- C. Moreau, R. Durand, C. Pourcheron and D. Tichit, Stud. Surf. Sci. Catal., 1997, 108, 399–406 CAS.
- W. Partenheimer and V. V. Grushin, Adv. Synth. Catal., 2001, 343, 102–111 CrossRef CAS.
- G. A. Halliday, R. J. Young and V. V. Grushin, Org. Lett., 2003, 5, 2003–2005 CrossRef CAS.
- C. Carlini, P. Patrono, A. M. R. Galletti, G. Sbrana and V. Zima, Appl. Catal., A, 2005, 289, 197–204 CrossRef CAS.
- O. C. Navarro, A. C. Canós and S. I. Chornet, Top. Catal., 2009, 52, 304–314 CrossRef.
- M. A. Lilga, R. T. Hallen and M. Gray, Top. Catal., 2010, 53, 1264–1269 CrossRef CAS.
- J. P. Ma, Z. T. Du, J. Xu, Q. H. Chu and Y. Pang, ChemSusChem, 2011, 4, 51–54 CrossRef CAS.
- A. Takagaki, M. Takahashi, S. Nishimura and K. Ebitani, ACS Catal., 2011, 1, 1562–1565 Search PubMed.
- B. Saha, S. Dutta and M. M. Abu-Omar, Catal. Sci. Technol., 2012, 2, 79–81 RSC.
- S. E. Davis, L. R. Houk, E. C. Tamargo, A. K. Datye and R. J. Davis, Catal. Today, 2011, 160, 55–60 CrossRef CAS.
- S. E. Davis, B. N. Zope and R. J. Davis, Green Chem., 2012, 14, 143–147 RSC.
- O. Casanova, S. Iborra and A. Corma, ChemSusChem, 2009, 2, 1138–1144 CrossRef CAS.
- Y. Y. Gorbanev, S. K. Klitgaard, J. M. Woodley, C. H. Christensen and A. Riisager, ChemSusChem, 2009, 2, 672–675 CrossRef CAS.
- T. Pasini, M. Piccinini, M. Blosi, R. Bonelli, S. Albonetti, N. Dimitratos, J. A. Lopez-Sanchez, M. Sankar, Q. He, C. J. Kiely, G. J. Hutchingsb and F. Cavania, Green Chem., 2011, 13, 2091–2099 RSC.
- N. K. Gupta, S. Nishimura, A. Takagakib and K. Ebitani, Green Chem., 2011, 13, 824–827 RSC.
- F. Koopman, N. Wierckx, J. H. Winde and H. J. Ruijssenaars, Bioresour. Technol., 2010, 101, 6291–6296 CrossRef CAS.
- M. L. Ribeiro and U. Schuchardt, Catal. Commun., 2003, 4, 83–86 CrossRef CAS.
- M. Kroger, U. Prüβe and K. D. Vorlop, Top. Catal., 2000, 13, 237–242 CrossRef CAS.
- G. A. Kraus and T. Guney, Green Chem., 2012, 14, 1593–1956 RSC.
- C. M. Lew, N. Rajabbeigi and M. Tsapatsis, Ind. Eng. Chem. Res., 2012, 51, 5364–5366 CAS.
| This journal is © The Royal Society of Chemistry 2012 |