Boron dipyrromethene (BODIPY)-based photosensitizers for photodynamic therapy†
Samuel G.
Awuah
ab and
Youngjae
You
*ab
aDepartment of Pharmaceutical Sciences, University of Oklahoma, Health Sciences Center, Oklahoma City, Oklahoma 73117, United States
bDepartment of Chemistry and Biochemistry, University of Oklahoma, Norman, Oklahoma 73019, United States. E-mail: youngjae-you@ouhsc.edu; Tel: +1 405 271 6593x47473
First published on 7th September 2012
Abstract
Photodynamic therapy (PDT) has shown promise as an effective treatment modality for cancers and other localized diseases, such as age-related macular degeneration and actinic keratosis. PDT relies on light, photosensitizer and oxygen as elements in its mechanism of action. BODIPY photosensitizers that have been under extensive study over the past decade have been demonstrated as a new class of photosensitizers. In this review, we attempt to summarize the decade-long study of BODIPY-based photosensitizers. We provide an overview of the superior photophysical properties possessed by BODIPY-based agents and the various synthetic strategies employed that have led to improved and selective and/or targeted photosensitizers.
 Samuel G. Awuah | Samuel G. Awuah received a B. Sc. from Kwame Nkrumah University of Science and Technology, Kumasi Ghana. He is presently completing a PhD at the University of Oklahoma in the area of Organic Chemistry with research focus in Medicinal Chemistry and NIR molecular probes for imaging and photodynamic therapy. His research interests are in the development of light harvesting materials for disease diagnosis and therapy, as well as solar cell applications. |
 Youngjae You | Dr Youngjae You is an associate professor at the Department of Pharmaceutical Sciences of the University of Oklahoma Health Sciences Center. He is also a member of the Peggy and Charles Stephenson Cancer Center. He obtained his PhD from Chungnam National University, Korea in 2001 and was a postdoctoral fellow and a research assistant professor at the SUNY Buffalo. He became an assistant professor in South Dakota State University and then moved to OUHSC in 2010. His interests are to develop NIR probes for cancer theranostics, light-controlled drug delivery systems and anticancer agents. |
A. Introduction
Photodynamic therapy (PDT) is an effective and approved regime for the treatment of certain types of cancers and precancerous inductions, age-related macular degeneration and actinic keratosis.1–4 It has also been explored for other diseases, such as localized infections, dermatological and cardiovascular illnesses and wound healing.5–7 Three elements are important components for effective photodynamic activity: light, oxygen and a photosensitizer (PS). BODIPY (4,4-difluoro-4-bora-3a,4a-diaza-s-indacene)-based and its aza analogue (Fig. 1) PSs (PSs) are a new class of PSs retaining characteristics of fluorescent BODIPY dyes such as excellent optical properties and synthetic versatility.8–10 This review seeks to summarize and discuss recent achievements in BODIPY-based PSs for the application of oncologic and antimicrobial PDT.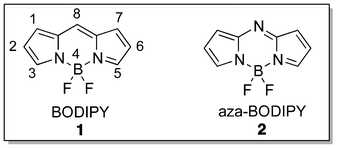 | ||
| Fig. 1 Chromophores of BODIPY and aza-BODIPY. | ||
B. Three key elements of PDT
PDT operates by a tripartite modality involving light, a PS and oxygen. A PS is administered and then the treatment site (e.g. tumor) is subjected to light irradiation to generate reactive oxygen species, especially singlet oxygen from oxygen, to damage target diseases. When a PS is irradiated, it is converted to the triplet state via intersystem crossing from the singlet state (Fig. 2).11 In what is known as the type II process, the triplet state PS transfers its energy to molecular oxygen to produce singlet oxygen. In a type I process, a chemical reaction of the excited PS with a substrate occurs, initiating an electron transfer leading to the formation of radicals, which react with molecular oxygen to produce reactive oxygen species, such as superoxide, hydrogen peroxide and hydroxyl ions.12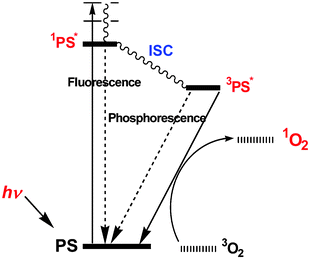 | ||
| Fig. 2 PDT action by a simplified Jablonski diagram. | ||
B1. Light
The use of light in photodynamic therapy has evolved significantly from the use of sunlight to very sophisticated light sources and light delivery tools with the aid of optical technology.4,13,14 Light for PDT can be classified into laser and non-laser light sources. Laser light is of a higher quality than the other light sources offering advantages such as high intensity, monochromaticity, no thermal effect by laser (400–800 nm) alone and easy adaptability with fiber optic delivery. Due to the attenuation of incident light by tissue, relatively high fluence rates are required to treat bulky targets. Monochromatic light makes the dosimetry more accurate than non-monochromatic light at least at the surface of the target area. Photofrin® and Visudyne® are approved in combination with laser irradiation (630 and 689 nm, respectively). However, traditional laser systems (e.g., argon/dye or KTP:YAG/dye) were not readily accessible due to high costs and large size. Diode lasers became more popular in the PDT community solving these problems to some degree. On the other hand, non-coherent light sources offer some advantages over lasers, such as low cost, wide range of spectrum, compact size, easier handling and less regulation. It is advantageous for dermatologic diseases and, in particular, less accurate dosimetric control may be acceptable. A narrow band fluorescence light source (BLU-U®, 400–450 nm) is used for Levulan for acne keratosis. The other type of fluorescence source is available for preclinical applications, such as Lumacare®, where specific filters are used to make narrow band outputs covering a wide range of the spectrum. Recently, light emitting diodes (LEDs) have become readily available for PDT application; they are portable and non-coherent, but adjustable to narrow ranges based on the semiconductor materials. The wavelengths are in the range of 350–1100 nm and can deliver a power output of up to 150 mW cm−2. PDT became more popular and accessible at least in part due to advances in light sources; the availability of good quality light, high power, low cost and customizable delivery using the fibers. It is important to mention that PDT thrives on light wavelengths with excitability in the popularly referred to “therapeutic window” (650 nm–800 nm) for deep tissue penetration. Therefore, designing a light source that excites at a longer wavelength is appropriate for non-invasive PDT treatment. Notable examples of such light sources are 690 nm and 785 nm diode lasers, as well as the variable long wavelength filters (>700 nm) of the Lumacare®.B2. Singlet oxygen – cytotoxic species
Singlet oxygen has been proven to be the main cytotoxic agent of PDT to cause biological effects among reactive oxygen species.11,15–18 The production of electronically excited two singlet oxygen states (1Σ+g (158 kJ mol−1) and 1Δg (95 kJ mol−1)) above the triplet state oxygen by the quenching of the lowest electronically excited singlet (S1) and triplet (T1) states of substances is possible if the transferred energy is higher than 158 kJ mol−1.19,20 The metastable, but reactive O2 (1Δg) can be generated by the deactivation of O2 (1Σ+g) through electronic and vibrational energy transfer.19,20 However, because the lifetime of T1 is much longer than S1, it is believed that singlet oxygen (1Δg) is generated by energy transfer from T1 to triplet oxygen.Three important aspects of singlet oxygen should be understood for analyzing biological data in PDT. First, the concentration of oxygen, the source of singlet oxygen, may impact on singlet oxygen generation capacity and consequently biological activity. At atmospheric pressure, oxygen concentration in water is about 0.27 mM.21 Each tissue has a different oxygenation status, e.g., pressures of oxygen are 40–100 mmHg in blood and 4–20 mmHg in tissues.22 In particular, solid tumors is known under a hypoxic condition.23 If oxygen concentration is reduced from 5% to 1%, it results in a 50% decrease in PDT effect.16 Secondly, the lifetime of singlet oxygen in cells was estimated to be short and singlet oxygen has a limited diffusion distance (10–300 nm).24–26 Thus, singlet oxygen generated in a cell may not directly damage neighboring cells via diffusion. It has pros and cons in biological implications. It gives PDT an opportunity for highly selective damage to target cells over non-targeted normal cells in the irradiated area. On the other hand, singlet oxygen cannot kill remaining live cells after PDT treatment in the irradiated area, which may arise from the heterogeneity of variables of tumor and PDT, such as PS distribution, oxygen concentration and light intensity.27–29 Thirdly, singlet oxygen quantum yield (ΦΔ) is the quantitative expression of a PS's ability to generate singlet oxygen.30,31 It has been used as one of the key parameters in assessing the potential of a PS for PDT, which seems reasonable in solution and in mono-layer culture models where the attenuation of the light is minimal. However, the attenuation of incident by tissue is huge.32 Almost 95% reduction of 690 nm light was observed for thin mouse skin without hair.33 Light becomes a major limiting factor in vivo. Thus, a more effective light observer should have a better chance to generate singlet oxygen with limited photons. A better term is needed for estimating the potential of a PS in vivo accounting for singlet oxygen generation quantum yield in combination with the extinction coefficient (ε) of the PS at the irradiation wavelength called “phototoxic power” (PP), where PP = ΦΔ × ε. Although a higher PP does not guarantee the effective in vivo PDT efficacy due to many other factors involved, it should be a better term than singlet oxygen quantum yield.
B3. Photosensitizers
PSs play a key role in PDT in that it is the drug that generates reactive oxygen species for cell damage upon light illumination. The report by Jesionek and Von Tappeiner in 1903 on the effect of the topical treatment of tumors with eosin exposed to white light marked one of the milestones in photodynamic events (Fig. 3).34,35 The extension of photodynamic activity to cancer was initiated by the observation of porphyrins in tumors by Polycard in 1924.36 Although several mouse model experiments corroborated the localization of haematoporphyrin and its analogues in tumors,37 it was not until 1960 that Richard Lipson and co-workers developed the “haematoporphyrin derivative” (HPD), which emitted fluorescence upon localizing in tumors.38–42 The treatment of mammary tumors in vivo with HPD and red light by Dougherty and his colleagues made a major transformation in photodynamic effects.43 Subsequently, Kelly and Snell performed the first human treatment with HPD for bladder cancer.44,45 Several preclinical and clinical trials of HPD and purified HPD led to its approval under the trade name Photofrin for photodynamic therapy treatment of Barrett's oesophagus, cervical cancer, endobroncheal cancer, oesaphageal cancer, gastric cancer and papillary bladder cancer in a number of countries. Photofrin induces phototoxicity through a cellular uptake mechanism, relying on intracellular oxygen levels. Photofrin-PDT cell death phenotype has been found to be largely dependent on the subcellular localization of Photofrin.46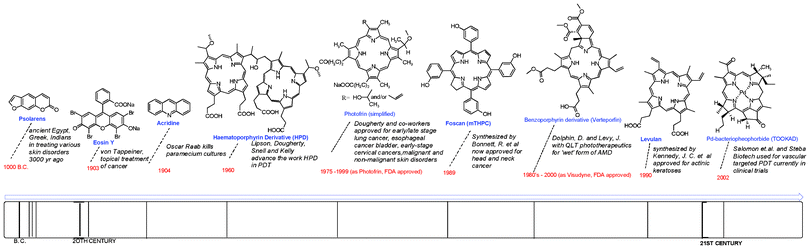 | ||
| Fig. 3 Milestones of photosensitizers in PDT. | ||
The development of efficient PSs led to the second-generation PSs to overcome the problems of Photofrin, such as chemical complexity, skin photosensitization and short tissue penetration due to absorption at 630 nm with a small extinction coefficient. The quest for more ideal PSs has opened the door to several porphyrin-based, as well as non-porphyrin-based PSs.47–53 Second-generation PSs satisfy fully or partially the following criteria: chemically defined structure (i.e. pure compound), effective singlet oxygen generation, absorption at photodynamic window (650–800 nm), low dark but high phototoxicity, good photostability and low skin photosensitization in vivo.
More recent PSs have been designed to achieve 1) absorption and emission near 800 nm with high efficiencies, 2) improved specificity to targets using targeting vectors or target-specific activation mechanisms and 3) dual modality for imaging and therapy.54–58 Stable bacteriochlorin-based PSs have been extensively studied for absorption near 800 nm with high extinction coefficient.54 Palladium-bacteriopheophorbide (TOOKAD), derived from the natural bacteriochlorophyl a, can be excited at 763 nm with a high extinction coefficient (∼100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1). Notably, it has been studied specifically for anti-vascular PDT while most of photosensitizers are known to induce three mechanisms: direct cell damage, immunolgical effect, and anti-vascular effect. TOOKAD, in fact, made a significant progress to clinical trials.59 Traditionally, PSs have been known to be specific to tumors with unknown mechanisms. However, improved delivery has been sought by using targeting vectors, such as ligands, peptides and antibodies.56,60–63 Active investigations have also been taking place for developing activatable PSs that can be transformed to the active form from the deactivated form by internal or external stimuli for improved target specificity.58 The unique procedure for PDT, i.e. light-activation, made it very attractive for developing theranostic applications: therapy and diagnostics.63,64 Various imaging modalities can be combined with PDT, such as MRI, fluorescence imaging, PET, etc. In particular, the combination of PDT and fluorescence imaging can offer a more effective management option for certain types of cancer.57,65–67
000 M−1 cm−1). Notably, it has been studied specifically for anti-vascular PDT while most of photosensitizers are known to induce three mechanisms: direct cell damage, immunolgical effect, and anti-vascular effect. TOOKAD, in fact, made a significant progress to clinical trials.59 Traditionally, PSs have been known to be specific to tumors with unknown mechanisms. However, improved delivery has been sought by using targeting vectors, such as ligands, peptides and antibodies.56,60–63 Active investigations have also been taking place for developing activatable PSs that can be transformed to the active form from the deactivated form by internal or external stimuli for improved target specificity.58 The unique procedure for PDT, i.e. light-activation, made it very attractive for developing theranostic applications: therapy and diagnostics.63,64 Various imaging modalities can be combined with PDT, such as MRI, fluorescence imaging, PET, etc. In particular, the combination of PDT and fluorescence imaging can offer a more effective management option for certain types of cancer.57,65–67
C. History of BODIPY-based photosensitizers
The BODIPY-based PSs are dipyrrole units connected by a methine (i.e. carbon atom at the meso position) or an aza-methine unit (i.e. nitrogen atom at the meso position) in a tricyclic fusion – a six membered ring with a boron center to constrain the structure flanked by two five-membered units. The Burgess group,8 as well as Ziessel et al.68 produce sterling information on the chemistry, synthesis, reactivity, chemical modification of BODIPY-based compounds and spectral details in their reviews. The boron dipyrromethene has inherent fluorescent properties with excellent optical properties, such as intense fluorescence quantum yields, sharp excitation and emission spectra, highly photo- and chemo-stablity69,70 and usablity in both lipophilic and aqueous media.67,68 The aza-BODIPY was first synthesized by Rogers in 1943 as a coloring agent,71,72 while in the late 1960s work by Treibs and Kruezer discovered the famous 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene (BODIPY).73 It was not until 2002 that O'Shea and co-workers published their findings on a structurally constrained aza-BODIPY and its application for photodynamic therapy. Their work gave an evidence of a new class of non-porphyrinic photodynamic therapy agent as they assessed the spectroscopic, photophysical and in vitro cellular uptake of the sensitizers.74 The group expanded their efforts to demonstrate the first in vitro and in vivo photodynamic action with a new class of aza-BODIPY PSs. In 2005, Nagano et al. showed the conversion of the highly fluorescent BODIPY into a potent PS with its excellent optical properties being maintained through the heavy atom effect.75 In 2009, O'Shea and co-workers published in vivo studies using aza-BODIPY-based PS.76D. Characteristics of the BODIPY compound
The BODIPY framework is known to be a versatile and robust fluorophore due to its numerous useful applications. In fact, the use of BODIPY for biological labeling via fluorescent conjugates of proteins, nucleotides, fatty acids and other biological units to understand biological systems and pathways makes it a photostable substitute for fluorescein and rhodamine among others.68 BODIPY can be tuned from wavelengths (excitation and emission) of about ∼500 nm to NIR wavelengths by structural modification with sharp absorption and emission bands. In addition to the flexible synthesis, several photophysical properties, such as singlet–triplet state character and electrochemical properties, can be tuned as desired. These properties have enabled the transformation of a fluorescent dye with negligible triplet state to a PS. A notable deficiency of most BODIPY PSs developed so far has been their excitability in the region of ∼500 nm, which renders them short of the therapeutic window (650–800 nm) for effective in vivo application. This has prompted the development of longer wavelength BODIPYs, as will be discussed later in this review.E. Structural modification of BODIPY-based photosensitizers and the heavy atom effect
E1. Halogenation and the heavy atom effect
The mesomeric structures of BODIPY show that its 2- and 6- positions are prone to electrophilic substitution, as typified by Treibs and Kreuzer in the use of chlorosulfonic acid for sulfonation.73 The electronic-rich property possessed by the BODIPY core makes it useful for chemical modification, especially the electrophilic aromatic substitution with halogens (Br, I), which converts the highly fluorescent or low triplet transition molecule to a potent PS with efficient intersystem crossing capacity due to spin orbit coupling by the heavy atom effect. The heavy atom effect may be described as the enhancement of a spin-forbidden process induced by the presence of an atom of a high atomic number, either internal or external to the excited molecule.77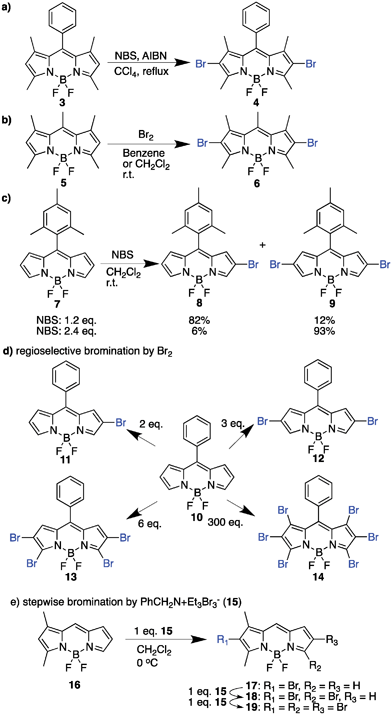 | ||
| Scheme 1 Bromination reactions of BODIPY. | ||
 | ||
| Scheme 2 Regioselective bromination of the pentacyclic BODIPY core. | ||
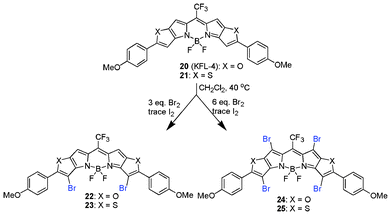
You et al., in an attempt to obtain long wavelength PSs, employed the bromination of highly conjugated BODIPY systems using bromine with trace iodine in CH2Cl2. This bromination approach was selective in generating di- (22 and 23) and tetra- (24 and 25) brominated forms.87
:1 mixture of chloroform/acetic acid at 25 °C for 10 h.89,90
E2. Short wavelength BODIPY Photosensitizer
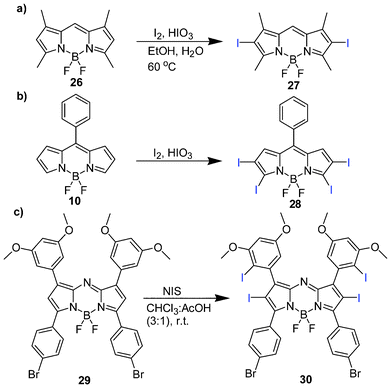
In this section, we focus on BODIPYs that absorb <600 nm, referred to as short wavelength PSs. The practical use of these PSs absorbing shorter wavelengths than the photodynamic window may be limited, especially for the treatment of solid tumors. Structural variations influence the efficacy of the BODIPY family of compounds for use as potent PSs. These short wavelength BODIPYs were synthesized in about 3–4 steps and maintained some of the original photophysical characteristics of its parent BODIPY fluorophore, such as absorption and emission maxima, sharp absorption and emission peaks. Early BODIPY-based PS development, like all other classes of PSs and general drug development, focused on generating efficient PSs with high singlet oxygen quantum yield before further modifications for improving biological activity.The iodination of the highly fluorescent 26 generates a PS (27) of singlet oxygen quantum yield ∼ 1 with exceptional photostable properties (Scheme 3a).75In vitro photosensitization of 27 was determined by loading HeLa cells with 1 μM for 30 min and irradiation with green light (535 ± 25 nm, 5 mW cm−2) for 1 min under a fluorescence microscope with a concurrent cell viability assay through the use of esterase and nucleic acid intercalating dyes, calcein AM and ethidium homodimer-1 (EthD-1). Phototoxicity was observed, proposing 27 to be a useful agent for PDT (Fig. 4). Phototoxicity of 27 against 3 different cell lines was also determined by another group: promyelocytic leukemia cell line (HL-60), oral squamous carcinoma cell line (HSC-2) and nasopharyngeal carcinoma cell line (HK1) following irradiation with 4.1 J cm−2 broadband light yielded IC50 values of 0.062 μM, 0.64 μM and 0.57 μM, respectively.91
 | ||
| Fig. 4 Cell photosensitization by 27 (2I-BDP). (A–C) Differential interference contrast (DIC) and fluorescence images of HeLa cells loaded with calcein AM (living cell marker) and EthD-1 (dead cell marker) after photosensitization with 27. (D–F) Loading with 27 alone had no toxic effect in this assay. Reprinted with permission from ref. 70. Copyright 2005 American Chemical Society. | ||
 | ||
| Scheme 3 Iodination of BODIPY and aza-BODIPY. | ||
Lee et al. investigated the structural implications on cell phototoxicity of 15 BODIPY analogues.91 The study involved functionalization by iodination at 2- and 6- positions, meso-substitution with iodophenyl and alkylcarboxylic acid, direct sulphonation at the 2,6-positions to improve the hydrophilicity and extended conjugation using fragments such as acrylic acid, ethenesulphonic acid and methyl acrylate. Among the four iodinated analogues (Fig. 5a), compound 34 shows potent photodynamic activity and is an attractive lead photochemotherapeutic agent owing to its facile synthesis with attendant incorporation of a carboxylic fragment for functionalization, which is useful in the attachment of targeting moieties. The synthesis relied on the incorporation of alkylcarboxylate functionality by the use of carboxylic anhydride in a one-pot process (Fig. 5b), as earlier reported by Wang et al.92 and Li et al.93
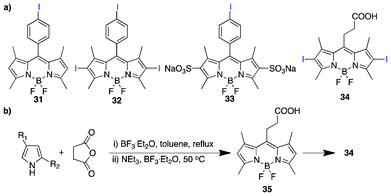 | ||
| Fig. 5 (a) Iodinated BODIPY PSs and (b) the synthetic scheme for 34. | ||
The intracellular accumulation of 34 loaded on the HSC-2 cell line as displayed in the co-staining of 34 and rhodamine 123 (mitochondria-specific dye) by spinning disk confocal microscopy exhibits particular localization in the mitochondria (Fig. 6a) over the endoplasmic reticulum and lysosome using ER tracker and Lysotracker, respectively (Fig. 6b and c). Compound 34 also induced apoptosis. In addition, an anti-angiogenic PDT effect was observed in the CAM assay (Fig. 6d). The well-vascularized chick chorioallantoic membrane (CAM) as the main respiratory organ of the chick embryo has shown to be an adaptive model for the studies of vascular damage in PDT.94,95 The ability of 34 to impose vasculature disruption using 3.5–7.0 nmol/embryo with a light dose of 20–40 J cm−2 is appreciable. The results show a dependence of vascular damage on drug-light doses with controls showing no damage. Since the anti-angiogenic effect is one of the three main mechanisms for anti-tumor PDT effects, along with direct cell killing and immunological effects, this compound has the potential to be a PS for anti-tumor PDT.
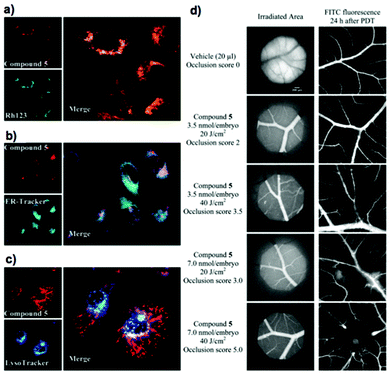 | ||
| Fig. 6 Intracellular localization of compound 34 (= compound 5 in (d)): spinning disk confocal images (a, b, c) of HSC-2 cells double-stained with 100 nM 34 and respective organelle probes. (a) Mitochondria (100 nM Rh123), (b) endoplasmic reticulum (100 nM ER-Tracker) and (c) lysosomes (500nM LysoTracker). Objective magnification is 63. (d) Representative angiographs of blood vessels supplying the CAM at the beginning and 24 h after PDT, illustrating the vascular occlusion efficacy induced by compound 5 at 3.5–7 nmol/embryo: irradiation at 510–560 nm of 20–40 J cm−2). Objective magnification is 4. Adopted with permission from ref. 84. Copyright 2010 American Chemical Society. | ||
E3. Long wavelength BODIPY-based photosensitizers
The development of BODIPY dyes absorbing/emitting in the far-red/near infrared region of the electromagnetic spectrum has attracted enormous research attention in recent years due to numerous advantages, especially in its biological application both in fluorescence imaging and PDT. Between 700–900 nm, absorption by cells/tissues and water, light scattering and autofluorescence is reduced significantly, allowing for deeper light penetration, resulting in efficient therapy and diagnosis.96 Thus, significant research effort has been made to generate BODIPY fluorophores absorbing at longer wavelength, >700 nm. It is important to mention that the strategies for BODIPY PSs absorbing in the far-red or NIR region rely heavily on its BODIPY fluorophore counterparts; hence the need to outline some of these fundamental approaches used for the fluorochromes. The following strategies can be used to induce bathochromic shift of BODIPY dyes (Fig. 7). 1) Aryl substitution at the 1,3,5 and 7-positions of the BODIPY core induce the red-shift.97,98 2) Vinyl substitution at the 2,6-positions with respective acrylate moieties (20–25) via palladium-catalyzed C–H functionalization in the Heck-type reaction99 red shifts by about 20–30 nm with monosubstitution and 50–60 nm with bis-substitution. Ethynylaryl substitution at the 3,5-positions of the BODIPY core is also an appealing approach to promote red shifting. The Sonogashira reaction using arylacetylene with its corresponding halogen-substituted BODIPY makes it possible.100,101 3) In some cases, the Liebeskind–Srogl coupling with boronic acids or tin-based compounds102 and nucleophilic substitution of halogen-substituted BODIPY at the 3,5-positions with conjugated groups induces red shifting.85,103 4) A popular approach used to date to obtain long-wavelength BODIPY PSs is the styryl substitution by Knoevanegel condensation. The versatility of the styryl substitution is unsurpassed104,105 in that it occurs in moderate yields and can be achieved in a stepwise fashion from monosubstituted to even tetrasubstitution with a corresponding red-shift.106 Akkaya and co-workers make a sterling observation in the assessment of the acidity of methyl protons in a tetramethyl-BODIPY, indicating electron densities in the order 2,6 ≫ 1,7 > 3,5.106 The methyl groups in positions 1 and 7 may be similar in acidity to that of 3,5 methyl groups. 5) Using aromatic ring fusion and extended aromatic systems is an intelligent approach to obtain long wavelength BODIPY dyes by extended conjugation. Chemical modification in this regard could be achieved via benzo-fusion following the retro-Diels–Alder reaction of norbornane-derived pyrroles by Ono and co workers.107–110 A facile method to obtain functionalized asymmetric benzo-fused BODIPY has been proposed by Hao et al.111 Heteroaryl fusion is attractive due to the bathochromic shift in the absorption and emission maxima it induces, as well as enhanced molar absorption. 6) In addition to the extensive π-system it imposes, it retains a rigidified and high planarity BODIPY chromophore, which directly affect the photophysical properties. Suzuki et al.70 and You et al.87 demonstrated the heteroaryl ring fusion using furan and thiophene at the C-2/C-3 and C-5/C-6 positions, respectively. 7) To further induce bathochromic shifting of absorption and emission maxima into the NIR region, incorporation of electron-donating (HOMO) and electron withdrawing (LUMO) moieties in a D–π–A fashion is useful – a common color chemistry phenomenon.112 8) The replacement of the meso-carbon with a N atom, as reflected in aza-BODIPY dyes, is a proven strategy for long-wavelength BODIPYs. The lone pair electrons on the nitrogen atom engineer a reduced HOMO–LUMO gap by interfering with the orbital architecture of the pyrroles, hence, the bathochromic shift.113 The aza-BODIPY analogues, ADPM06, in PDT have shown great promise with their far-red absorption being a strong selling point. 9) It is worth noting that constraining or rigidifying rotatory moieties can also induce red shifting.74,114 Overall, an extended π-system with simultaneous retention of the molecule's rigidity and increased planarity promote long-wavelength BODIPY dyes. A highly functionalized, water-soluble long-wavelength BODIPY PS still remains a challenge to circumvent.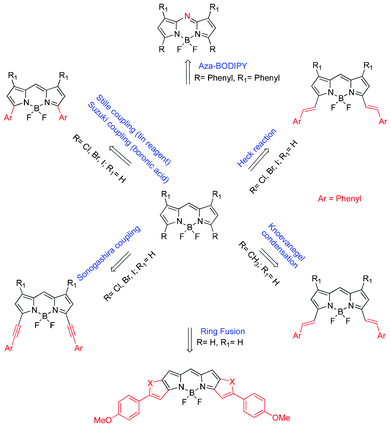 | ||
| Fig. 7 A schematic summary of chemical modification for long-wavelength BODIPY dyes. | ||
E3.1. Styryl/Distyryl BODIPY-based photosensitizers
The styryl group incorporation provides a red-shift to approximately 100 nm (i.e. ∼600 nm absorption maxima)115 with further red shifting obtained with the introduction of a second styryl group (i.e. ∼680 nm).113 The styryl incorporation by the Knoevanegel condensation into the BODIPY core can be achieved symmetrically and/or unsymmetrically without compromising the excellent photophysical properties possessed by BODIPY dyes. The introduction of functionalized styryl and distyryl groups with water-soluble moieties, such as pegylated structures, enhanced biocompatibility, hydrophilicity and cellular uptake. Akkaya et al.116 and Ng et al.117 showed the in vitro localization and photocytotoxicity of water-soluble and symmetrical distyryl BODIPY PSs (Fig. 8a and b).116,117 The PS design possessed a heavy atom (bromine/iodine) substituted at the 2,6-positions to facilitate intersystem crossing as usual and the introduction of triethylene glycol subunits to improve water solubility, which is a well precedented phenomenon to enhance cell permeability and tumor targeting properties in PSs118–120. PS 38 absorbs maximally at 660 nm with a high extinction coefficient of 102000 M−1 cm−1, with no sign of aggregation in aqueous buffer medium. The phototoxicity with LED irradiation (625 nm at 2.5 mW cm−2, 4 h → 36 J cm−2) showed an EC50 of less than 200 nM. PS 41 also showed a very low IC50 (7 nM) against HT29 cells following irradiation (>610 nm, 40 mW cm−2, 20 min → 48 J cm−2), with low aggregation in media.
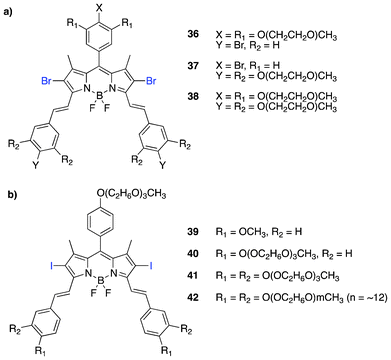 | ||
| Fig. 8 Structures of dihalogenated symmetric distyryl BODIPY PSs. | ||
Asymmetrical distyryl BODIPY derivatives provide varying functionalization options to the applicability in PDT; they could offer the introduction of water-soluble moieties, such as polyethylene glycol units, as well as red-shifting through extended conjugation. Ng et al. prepared diiodinated asymmetric distyryl analogues (Scheme 4).121 The chemical modification to achieve unsymmetrical distyryl BODIPY PSs is straightforward. Treatment of diiodo BODIPY with two different aryl aldehydes sequentially in 1 equivalence affords the variant condensed BODIPY. PS 47 showed high phototoxicity: IC50 of 15 nM after irradiation (>610 nm, 40 mW cm−2, 20 min → 48 J cm−2).
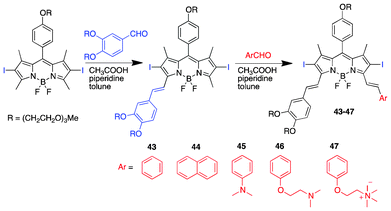 | ||
| Scheme 4 Structures and synthesis of diiodinated asymmetric distyryl BODIPY PSs. | ||
Akkaya et al. proposed that SWNT (singlet wall carbon nanotube) has the potential to be used for delivery of PSs using non-covalent links.122 Pyrenyl-functionalized distyryl-BODIPY (48) was non-covalently added to SWNT. Singlet oxygen generation by indirect DPBF oxidation experiments showed 25% degradation of DPBF within 40 min with a result using a low concentration of conjugate 48–SWNT (62.0 nM) as compared to just 48 (50 μM).
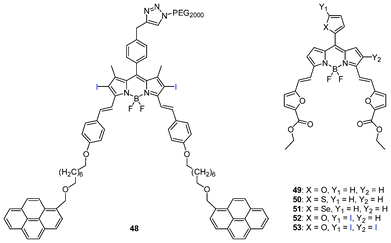 | ||
| Fig. 9 Pyrenyl-functionalized distyryl and heavy atom-substituted difuranylvinyl-BODIPYs. | ||
Prasad and co-workers prepared a new class of distyryl BODIPY PSs (Fig. 9), e.g., 3,5-difuranylvinyl-BODIPYs (49–53).123 These BODIPYs can be synthesized by building a styryl-derived pyrrole possessing a substituted phenyl or heteroatomic subunits through the Wittig-type reaction. The approach generated PSs that absorb and emit in the far-red and NIR region (λabs = 628–713 nm and λem = 641–746 nm) with significant generation of 1O2 proved by the direct luminescence method with a phosphorescence spectra at 1270 nm from 1O2.
E3.2. Aza-BODIPY photosensitizers
The aza-BODIPY PSs prepared by O'Shea et al. are the most successful thus far in PDT among BODIPY-based PSs, owing to their excitation in the far-red region (∼650–688 nm), high generation of 1O2 (ΦΔ = 0.74) and excellent optical and photophysical properties, such as a molar extinction coefficient of 75000 M−1 cm−1 and relatively good emission (Φf = 0.1), with a PP of 55500 M−1 cm−1.84 The BF2 chelation of 3,5-diaryl-1H-pyrrol-2-yl-3,5-diarylpyrrol-2-ylidene amines (tetraarylazadipyrromethenes) with subsequent incorporation of heavy atoms, such as bromine, to increase the triplet state population – a requisite for singlet oxygen generation – introduces a new class of photodynamic therapeutic agents.74
Structural modifications of the aza-BODIPYs displayed varying degrees of singlet oxygen generation based on heavy atom (bromine) substitution without significant distortion to the planarity of the PSs (Fig. 10).83 The positioning and number of heavy atom substitution modulated the generation of 1O2 due to spin–orbit perturbation.124 To prove the possibility of spin–orbit perturbation, O'Shea and co-workers designed three aza-BODIPYs according to the following modules: i) relying on the inherent spin–orbit coupling of the aza-BODIPY without heavy atoms; ii) intramolecular external heavy-atom effect125–128 in which bromine atoms are positioned on the aryl rings and not on the aza-BODIPY core, giving rise to moderate 1O2 generation and iii) internal heavy atom effect129–131 in which there is direct substitution of bromine atoms on the aza-BODIPY, yielding the 1O2 efficiently.
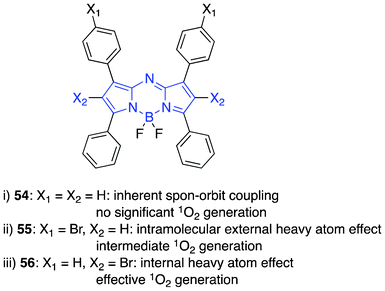 | ||
| Fig. 10 Singlet oxygen modulation by bromine positioning on the sensitizer core. | ||
Phototoxicity against HeLa and MRC5-SV40 (transformed fibroblast) cell lines validated 1O2 generation in vitro; the most attractive module being the bromine substituted directly on the sensitizer to produce an internal heavy atom effect. The potential PDT-drugs were formulated using cremophor and displayed perinuclear localization. Irradiation of the plates was achieved using a long-wavelength light source 600–750 nm at a light dose of 8 or 16 J cm−2 with the most effective (58, ADPM06, Fig. 11) showing an EC50 of 63 nM and 37 nM at 8 J cm−2 with no determinable dark toxicity in HeLa and MRC5-SV40 cell lines, respectively.
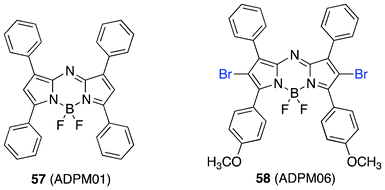 | ||
| Fig. 11 Structures of ADPM01 and ADPM06. | ||
Further development of the aza-BODIPYs as effective anti-cancer therapeutics required comprehensive studies on ADPM06.132 The lead compound displayed localization in the cytoplasmic compartment with a large accumulation in the endoplasmic reticulum with minor localization to the mitochondria. ADPM06 induced apoptosis as a mechanism for cell death. Interestingly, under hypoxic conditions (1% oxygen) ADPM06 shows significant phototoxicity at 16 J cm−2, with EC50 units of 1.5 and 1.6 μM for HeLa and MRC5 cell lines, respectively.
The in vivo efficacy and mechanism of action of ADPM06 corroborated the anticancer therapeutic capacity of the BF2 chelated aza-BODIPY.76 In the tolerance study using LLC/1 tumour-bearing mice, ADPM06 showed early signs of tumor response. A critical look at the toxicity profiles revealed mass necrosis within 72–96 h at 150 J cm−2. Oedema and inflammation was observed by 1–2 days post-PDT treatment, with the oedema healing in a few days without tissue damage to the surrounding normal tissue. However, tumors took approximately 20–30 days to heal. Studies on xenograft models were carried out using MDA-MB-231-GFP cells inoculated subcutaneously into nude mice. Tumor sizes of 7–9 mm diameter were raised within 7–10 days for PDT treatment. Ablation of tumor was achieved with drug-light doses of 2 mg kg−1 ADPM06 + 100–200 J cm−2 (Fig. 12a). The highest cure rate of 71% was achieved at a drug-light dose of 2 mg kg−1 ADPM06 + 150 J cm−2 light.
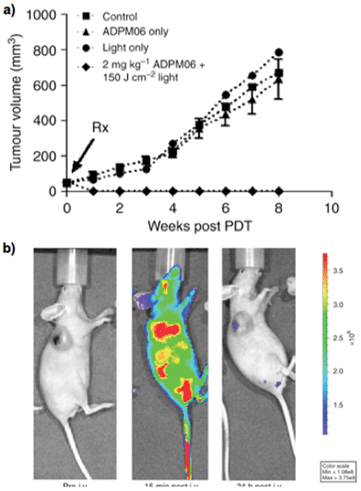 | ||
| Fig. 12 (a) In vivo response to ADPM06-mediated PDT against MDA-MB-231-GFP xenograft model: PDT group (2 mg kg−1 ADPM06 IV + 150 J cm−2 light (n = 7)), light-alone group (150 J cm−2 (n = 8)), drug-alone group (2 mg kg−1 ADPM06 i. v. (n = 14)) and vehicle control (n = 9). (b) In vivo fluorescence images of ADPM06, as measured by optical imaging: Balb/C nude mice bearing subcutaneous MDA-MB-231-luc tumours before and after i. v. injection of ADPM06 (2 mg kg−1). Adopted with permission from ref. 71. Copyright 2009 Nature Publishing Group. | ||
One notable point with ADPM06 is that optical imaging could be used to establish drug biodistribution and clearance by ex vivo fluorescence imaging from excised organs (Fig. 12b), which has been difficult with porphyrin-based PSs due to low fluorescence yield. It has been believed that one chromophore cannot be suitable for both singlet oxygen generation and fluorescence emission because these processes are competing. In other words, one chromophore cannot have both high singlet oxygen quantum yield and fluorescence quantum yield, which is true. However, it is possible to make azaBODIPYs with sufficient fluorescence brightness and high phototoxic power by balancing fluorescence and singlet oxygen quantum yields. For example, ADPM06 has achieved the correct balance to achieve both fluorescence imaging and high phototoxicity with a fluorescence quantum yield of 0.1 and a singlet oxygen quantum yield of 0.74.62,67,84
Anti-vascular PDT is effective since blood oxygen concentrations are high at vascular sites with high PS levels in blood shortly post-injection. In addition, the accessibility of vascular endothelial cells to PS molecules in blood is facile and direct. These benefits promote rapid vascular occlusion, discharging the PS molecules into the tumor region for efficient PDT.133,134 The vascular targeting effect of ADPM06 was demonstrated by MRI and positron emission tomography in vivo using a short drug-light interval. After 18FDG-PET evidence showing transient metabolic status, i. v. injection of Gd-DTPA contrast agent (0.6 ml min−1) and MRI scans ∼3, 24 and 48 h post-treatment display a reduced vascular perfusion (Fig. 13).
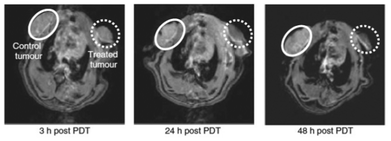 | ||
| Fig. 13 MRI approach to assessing vascular-targeting effect in vivo. The MRI of control 13762 MAT B III rat tumour (white circle) and PDT-treated tumour (0.8 mg kg−1 ADPM06 150 J cm−2 light) (dashed white circle) at 3, 24 and 48 h post-treatment with a Gd-DTPA injection, illustrating the change in tumour perfusion. Adopted with permission from ref. 71. Copyright 2009 Nature Publishing Group. | ||
ADPM06-mediated PDT at short drug-light intervals imparts complete response rates in a variety of cancer models. An apoptosis response initiated by ROS beginning in the ER post-PDT with concurrent caspase activation in vitro can also be observed.135
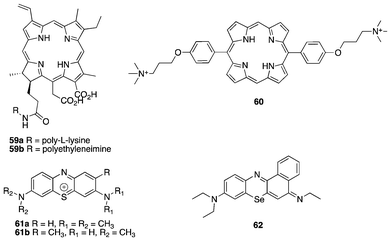 | ||
| Fig. 14 Antibacterial PDT agents. | ||
In recent developments, Reddy et al. used a porphyrin–apidaecin conjugate as a new broad spectrum antibacterial agent, underscoring the use of cationic moieties in antibacterial PDT.142 Porphyrins substituted at the meso (60) positions with ammonium salts perform photoinactivation of gram-positive bacteria to a large extent with reduced toxicity on eukaryotic cells.143 To further affirm the efficacy of antimicrobial PDT, the use of PSs in the phenthiazinium class, such as methylene blue (61a) and toluidine blue (61b), exhibit potency against blood pathogens and, in the case of methylene blue, decontaminate freshly frozen blood plasma.144,145 Like many PSs where the heavy atom effect contributes largely to ISC and attendant singlet oxygen generation. 62 containing selenium displays a far significant photoinactivation to a wide range of pathogens.146
To obtain a single agent for antimicrobial PDT with a more straightforward synthesis amenable to structural modification with excellent photophysical properties, the aza-BODIPY was exploited. Drawing insights from ADPM06,83 O'Shea et al. synthesized a PS bearing two cationic ammonium salts, 63 (Fig. 15).147
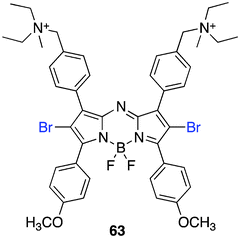 | ||
| Fig. 15 Aza-BODIPY antibacterial PS. | ||
The synthesis begins with the already reported bis-amine azadipyrromethene that can be accessed in three steps from 4-diethylaminomethyl benzaldehyde and 4-methoxyacetophenone. Bromination at room temperature and BF2 chelation with boron trifluoride and diisopropylethylamine were followed by alkyl substitution with iodomethane, which affords the target compound.
The photodynamic applicability of 63 in inactivating microbes is broad. Experimentally, the PSs with the bis-ammonium salts facilitates rapid uptake, ∼10 min in Gram-positive and negative bacterial strains and pathogenic yeasts which is due to the amphiphilic nature of the PS and the log P values close to zero (−0.25). Confocal images of 63 confirmed the broad-spectrum microbial uptake after a 10 min incubation period. A localization pattern observed in E. coli showed staining throughout the cell with an intense staining pattern on the outer periphery edge of the cell. Antibacterial phototoxicity using 1, 2 and 5 μg mL−1 of 63 was significant. A notable drug-light dose of 5 μg mL−1 and 16 J cm−2 achieved a 6.8 and 3.4 log10 reduction of S. aureus and a methicillin-resistant S. aureus (MRSA) strain, respectively. Additionally, using 5 μg mL−1 of PS and 75 J cm−2 a broad pathogenic inactivity resulted up to 3.6 and 5.7 log10 for gram-negative bacterium E. coli and the pathogenic yeast C. albicans, respectively.
E4. Heavy atom-free BODIPY photosensitizers
Developing BODIPY photosensitizers without the incorporation of heavy atoms, such as iodine, bromine, selenium, sulphur and certain lanthanides, may offer reduced dark toxicity. The low triplet population of the BODIPY chromophore means a focus on the excited state properties may be the key to achieving high ISC without heavy atoms. The recent developments of heavy atom-free BODIPY PSs has focused on: 1) dimeric BODIPY at an orthogonal orientation, which imparts an unperturbed HOMO → LUMO due to the lack of orthogonal π-framework mixing (a thorough discussion on theoretical insight is given in section H of this review);148 or 2) the use of spin convertors, such as C60, in dyads to sensitize photophysical processes, like singlet oxygen generation, by intramolecular energy transfer (EnT). The former employs the synthesis of meso-β linked BODIPY with visible absorption of 500–542 nm and ΦΔ = 0.21–0.51.148,149 The first approach resulted in a potent photosensitizer with in vitro phototoxicity at nanomolar concentrations, but it looks far from practical for in vivo use in PDT due to the short wavelength (∼520 nm). The latter strategy uses the light harvesting styryl-BODIPY or azaBODIPY150 in conjugation with C60 units. Their photophysical properties are attractive, with absorption at 629–657 nm and a high ΦΔ of 0.85 (for styryl BODIPY). The translation of these dyads into biological efficacy would be interesting.151F. Activatable BODIPY-based photosensitizers
Activatable PS is a unique approach to achieve additional selectivity in PDT to diseased tissue over normal tissue.58 Activatable BODIPY-based fluorophores have been successfully demonstrated in imaging to distinguish healthy cells from diseased ones.152–154 Since the singlet oxygen generation shares some dynamic processes with fluorescence emission, the activation mechanisms for BODIPY-based PSs may be adopted from those for BODIPY fluorophores. There have been a number of activation modalities for BODIPY-PSs that have been studied over the years, such as pH, hydrophobicity, nucleic acid activated and ions, most of which control photo-induced electron transfer (PeT, Fig. 16).PeT is a well-studied on/off switch mechanism to control fluorescence.155 The selective photoinactivation of specific proteins, also called CALI (chromophore assisted light inactivation), using the PeT approach in combination with environment activation strategies was demonstrated (Fig. 17b).156 The development of PSs bearing varying electron donors as a tunable PeT quencher with an inositol 1,4,5-trisphosphate receptor (IP3R) ligand presents effective on/off switching of 1O2 generation. The transfer of electrons by the donor moiety quenches the 1O2 generation (switch off) of the unbound molecule. However, upon specific binding to the hydrophobic pocket of the inositol 1,4,5-trisphosphate receptor, the switch is on, ready to generate 1O2 upon irradiation. This PeT-hydrophobic environment strategy demonstrated a unique activatable PS approach based on the BODIPY chromophore for the selective inactivation of protein function.
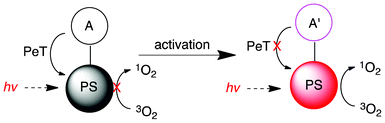 | ||
| Fig. 16 The general mechanism of PeT. PS: photosensitizer, A: auxiliary group, A′; modified auxiliary group. | ||
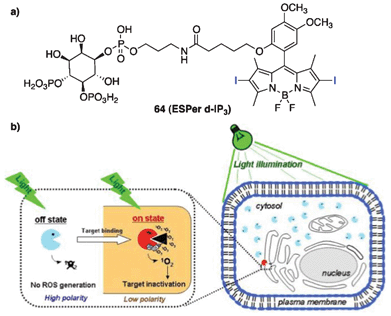 | ||
| Fig. 17 (a) The structure of 64 (ESPer d-IP3) and (b) a schematic representation of protein photoinactivation using ESPers. In the cytosolic polar environment, ESPers are in the off state of photosensitization (indicated by light blue) and do not generate 1O2. In the hydrophobic environment of the target, ESPers are in the on state of photosensitization (indicated by red) and generate 1O2, inducing inactivation of the nearby protein of interest. Adopted with permission from ref. 142. Copyright 2008 National Academy of Sciences, USA. | ||
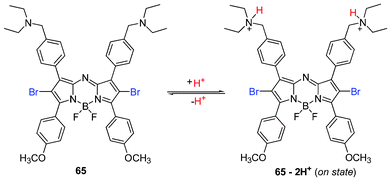
PeT was also utilized as a selective mechanism for ADPM PSs bearing amine receptors activated by pH. In this approach, a proton source acted as an activator, so called supramolecular photonic therapeutic agents (SPTA).157 The approach took advantage of the lower pH (6.5–6.8) associated with the interstitial fluid surrounding the tumor mass over normal tissue. Different amine receptors can modulate the degree of 1O2 generated, thereby impacting the phototoxicity. The on/off switch by the PeT–pH response strategy utilized the rationally chosen amine receptors to achieve significant cytotoxicity of EC50 values of up to 5.8 nM at 16 J cm−2 light irradiation with 65.
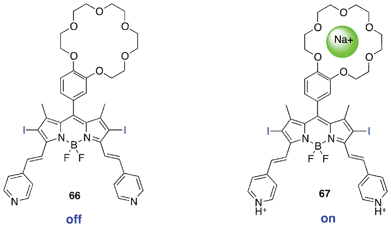
Molecular logic gates were proposed for use in synthetic RNA-based logic devices (fluorescent proteins),158 small object coding159 and pro-drug activation,160 to mention a few examples. Designs involving a receptor (molecule) with two input channels mimic the molecular AND logic gate approach to achieve selectivity.161 The AND gate is designed to follow a digital logic gate system, in which a logical conjunction of all high inputs to the AND gate yields a high output and a low output is obtained if neither or only one input is high.161,162 BODIPY dyes present a wide range of synthetic flexibility to obtain a “chemical” AND logic gate. Singlet oxygen acts as the output with chemical inputs drawn from the microenvironment of the diseased tissue, such as cancers. Large acidic vesicles other than endosomes and lysosomes contained in metastatic cancer cells may contribute to a the low pH of <4.163 In addition, intracellular sodium ion concentration is ∼3 times higher than in normal tissues.164 The variants observed in the cancer tissue environment make Na+ and H+ unique chemical inputs for the BODIPY AND logic gate.165 The smart synthetic design possesses a crown-ether-based PeT modulator sensitive to only Na+ and dimethylaminostyryl or pyridilethynyl fragments sensitive to acidic treatment (H+) (Fig. 18). Therefore, following the principle of molecular AND logic gates, high concentrations of both Na+ and H+ ions turns on the output, producing singlet oxygen in significantly high amounts relative to neither or only one high Na+ or H+. Although this application may not have been tested in vitro, it offers a potentially selective approach to the treatment of malignant diseases by molecular-based logic gates.
 | ||
| Fig. 18 Structures of aza-BODIPY PSs. | ||
G. Computational platforms of BODIPY-based photosensitizers
Computational calculations are not perfect yet, but are useful for various applications in the drug design process to save time and other resources. Theoretical calculations of BODIPY-based PSs have also been proven useful in the design of new PSs and analysing experimental data. In particular, quantum mechanical calculations were used to calculate geometry and electronic characteristics of molecules, such as geometry optimization, electronic spectra, energy levels and gaps, and electron density maps, which are important determinants for key photophysical properties of effective PSs.A detailed computational study on the tetraazadipyrromethene class of dyes developed by Russo et al. revealed the geometric character, excited state behaviour (both absorption band and singlet–triplet energy gap) and solvent effect providing useful insights into these novel classes of PSs.166 The computational model used was the Gaussian 03 package employing DFT level and PBE0 hybrid functional with 25% of exact exchange in association with split valence basis sets supported with polarization functions (SVP). The 6-31+G* and the PBE0 functional was appropriate in the prediction of the absorption bands by TD-DFT calculations, while the polarization continuum model (PCM) model was suitable in assessing the solvent effects.
The positioning of the four phenyl rings in relation to the core of the aza-BODIPY structure is defined by the four dihedral angles (Ψ1, Ψ2, Ψ3, Ψ4) that describe the conformation of the ground state geometric structures (Fig. 20). Thus, the most stable conformers were calculated relative to the dihedral angles. The calculated bond lengths and balance angles of the optimized structures were in agreement with experimental data. The four phenyl rings were positioned out of plane at the energy minimized conformations. Theoretical predictions of the absorption band were also compared with experimentally observed UV-visible spectra, also showing impacts of the substituents on the absorption maxima. A more interesting and useful prediction was the singlet–triplet energy gap, since a fundamental requisite for PSs to obtain PDT efficacy is displayed by its singlet–triplet energy gap (≥0.98 eV). However, some differences seen in the computational assessment warrants a caution in the interpretation of energy gap results for predicting singlet oxygen generation capability.
 | ||
| Fig. 19 BODIPY-based molecular AND logic gate. | ||
You et al. in an effort to understand the theoretical framework of NIR BODIPY dyes (20–25, Scheme 2) that generate singlet oxygen used the density functional theory (DFT) and time-dependent density functional theory (TDDFT) in Gaussian 09 to gain an insight in to the geometric and excited state character of the synthesized compounds.87 Using the 6-311G* basis set for geometry optimization and the PBEPBE correlation functional in vacuo, the excited state calculation exhibited strong absorption bands in the NIR region, which compared appropriately with experimental UV spectra data at ΔE = 0.06 eV (Fig. 21).
 | ||
| Fig. 20 The structure of BF2-chelated tetraarylazadipyrromethenes used in the study, with the atom, substituent and free-rotation torsion angle labelling. | ||
Ortiz et al. used electrostatic potential maps and Mulliken charges of BODIPY to estimate sites for electrophilic aromatic substitution in the polyiodination of BODIPY.88 The calculated data were used to rationally justify the sites of iodination, which were not only affected by an electronic factor, but also by a steric factor. They also observed that photophysical properties of polyiodinated BODIPYs depended on the sites of iodination. Iodination at the 3 or 5-position improved the absorption and emission efficiencies more than the other positions.
Recently, Akkaya and co-workers nicely proved a new concept for generating singlet oxygen from BODIPY dyes without using heavy atom effects.148 The heavy atom-free BODIPY PSs have been further explored through styryl BODIPY-C60 dyads showing strong absorption of visible light at 657 nm and an extinction coefficient of 64600 M−1 cm−1 with a delay of ΦΔ = 0.85. The concept was supported by theoretical insights into excited states using the complete active space SCF (a variant of MCSCF) with large basis sets and different active spaces. They synthesized orthogonal BODIPY dimers as a non-halogenated BODIPY efficient PS, where the tuning of excited states properties by raising the structure and electronic compatibility of S1 and T1 states over S1–S0 (fluorescence pathway) increased intersystem crossing without the use of heavy atoms. Singlet oxygen was directly detected using a near-IR sensitive detector and potent phototoxicity (EC50 = 50 nM) was observed against K562 human erythroleukemia cells after irradiation (520 nm LED, 2.5 mW cm−2, 4 h). From more extensive theoretical and experimental studies, they found that DS-TR (doubly substituted tetraradical configuration) is a valuable descriptor for estimating the photosensitization potential of such orthogonal dimers.167 However, the orthogonal BODIPY dimers may not be good for pursuing longer wavelength absorbing PSs. The extension of the π-system of BODIPY in the dimers seemed to destabilize the DS-TR, but stabilize SS (Fig. 22). The dimers with π-extended conjugated BODIPY did not generate singlet oxygen. This is an excellent case of using computer-assisted excited PS design and getting an insight into the excited states to understand behaviours of PSs.
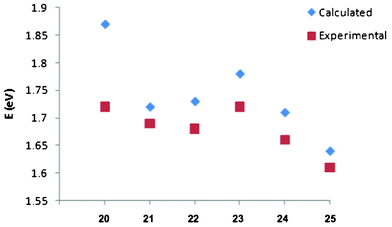 | ||
| Fig. 21 The observed trend for calculated excitation energies and experimental absorption maxima in eV. Adopted with permission from ref. 81. Copyright 2011 American Chemical Society. | ||
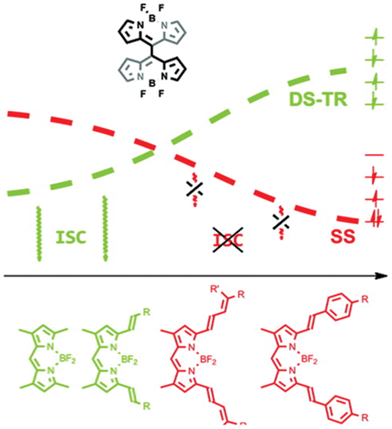 | ||
| Fig. 22 Relation of DS-TR and SS characters of S1 to pi-conjugation and tendency of ISC (DS-TR: doubly substituted tetraradical S1 state, SS: singly substituted HOMO → LUMO type open shell S1). Top: configuration of dimers, Bottom: monomeric structures of the dimers. Adopted with permission from ref. 152. Copyright 2012 American Chemical Society. | ||
H. Conclusion and future direction
Since the early publicaton by O'Shea et al. of aza-BODIPY PSs in 2002, the BODIPY class PSs have shown great promise in a very short period of time. ADPM06 is an excellent example that was demonstrated to be effective for in vivo models. It is unequivocal that BODIPY-based PSs have great potential as a new class of PSs, offering critical advantages, such as the excellent and tunable photophysical properties and flexible synthesis. Those make BODIPY a very attractive chromophore for an ideal PS. In addition, the recent explosion in knowledge about BODIPYs as fluorescence dyes, simple methods for converting from fluorescence dyes to PSs and relatively reliable theoretical tools could provide ideas for designing new BODIPY-based PSs. As demonstrated by the presented cases, BODIPY-based PSs can be used as an excellent platform for advanced PSs with 1) absorption and emission near 700 nm with high efficiency, 2) activatable photosensitization and 3) a dual functionality for theranostic PSs, balanced and effective singlet oxygen generation and fluorescence emission. Based on such promising results at very early drug discovery stages, BODIPY-based PSs should be proved effective at more advanced discovery stages, such as preclinical and eventually clinical studies.Acknowledgements
We acknowledge the Oklahoma Center for the Advancement of Science and Technology (OCAST) for financial support and ChemAxon for free access to Instant JChem used in compiling the BODIPY database found in the ESI†.References
- T. J. Dougherty, C. J. Gomer, B. W. Henderson, G. Jori, D. Kessel, M. Korbelik, J. Moan and Q. Peng, J. Natl. Cancer Inst., 1998, 90, 889–905 CrossRef CAS.
- M. A. MacCormack, Semin. Cutaneous Med. Surg., 2008, 27, 52–62 CrossRef CAS.
- T. J. Dougherty, J. Clin. Laser Med. Surg., 2002, 20, 3–7 Search PubMed.
- A. Juzeniene, Q. Peng and J. Moan, Photochem. Photobiol. Sci., 2007, 6, 1234–1245 CAS.
- P. Babilas, S. Schreml, M. Landthaler and R. M. Szeimies, Photodermatol., Photoimmunol. Photomed., 2010, 26, 118–132 CrossRef CAS.
- S. Kossodo and G. M. LaMuraglia, Am. J. Cardiovasc. Drugs, 2001, 1, 15–21 CrossRef CAS.
- J. Garrier, L. Bezdetnaya, C. Barlier, S. Grafe, F. Guillemin and M. A. D'Hallewin, Photodiagn. Photodyn. Ther., 2011, 8, 321–327 CrossRef CAS.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS.
- N. Boens, V. Leen and W. Dehaen, Chem. Soc. Rev., 2012, 41, 1130–1172 RSC.
- G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184–1201 CrossRef CAS.
- K. Plaetzer, B. Krammer, J. Berlanda, F. Berr and T. Kiesslich, Lasers Med. Sci., 2009, 24, 259–268 CrossRef CAS.
- C. S. Foote, Photochem. Photobiol., 1991, 54, 659 CrossRef CAS.
- L. Brancaleon and H. Moseley, Lasers Med. Sci., 2002, 17, 173–186 CrossRef CAS.
- T. S. Mang, Photodiagn. Photodyn. Ther., 2004, 1, 43–48 CrossRef.
- K. R. Weishaupt, C. J. Gomer and T. J. Dougherty, Cancer Res., 1976, 36, 2326–2329 CAS.
- J. Moan and S. Sommer, Cancer Res., 1985, 45, 1608–1610 CAS.
- H. I. Pass, J. Natl. Cancer Inst., 1993, 85, 443–456 CrossRef CAS.
- B. W. Henderson and T. J. Dougherty, Photochem. Photobiol., 1992, 55, 145–157 CrossRef CAS.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351–371 CrossRef CAS.
- R. Schmidt, Photochem. Photobiol., 2006, 82, 1161–1177 CrossRef CAS.
- M. Montalti and S. L. Murov, Handbook of Photochemistry, CRC/Taylor & Francis, Boca Raton, 2006 Search PubMed.
- M. Sitkovsky and D. Lukashev, Nat. Rev. Immunol., 2005, 5, 712–721 CrossRef CAS.
- J. M. Brown and W. R. Wilson, Nat. Rev. Cancer, 2004, 4, 437–447 CrossRef CAS.
- J. Moan, J. Photochem. Photobiol., B, 1990, 6, 343–344 CrossRef CAS.
- M. K. Kuimova, G. Yahioglu and P. R. Ogilby, J. Am. Chem. Soc., 2009, 131, 332–340 CrossRef CAS.
- M. Niedre, M. S. Patterson and B. C. Wilson, Photochem. Photobiol., 2002, 75, 382–391 CrossRef CAS.
- S. M. Hahn, M. E. Putt, J. Metz, D. B. Shin, E. Rickter, C. Menon, D. Smith, E. Glatstein, D. L. Fraker and T. M. Busch, Clin. Cancer Res., 2006, 12, 5464–5470 CrossRef CAS.
- T. M. Busch, S. M. Hahn, E. P. Wileyto, C. J. Koch, D. L. Fraker, P. Zhang, M. Putt, K. Gleason, D. B. Shin, M. J. Emanuele, K. Jenkins, E. Glatstein and S. M. Evans, Clin. Cancer Res., 2004, 10, 4630–4638 CrossRef CAS.
- T. M. Busch, E. P. Wileyto, M. J. Emanuele, F. Del Piero, L. Marconato, E. Glatstein and C. J. Koch, Cancer Res., 2002, 62, 7273–7279 CAS.
- F. Wilkinson, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1995, 24, 663–1021 CrossRef CAS.
- F. Wilkinson, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1993, 22, 113–262 CrossRef CAS.
- P. Juzenas, A. Juzeniene, O. Kaalhus, V. Iani and J. Moan, Photochem. Photobiol. Sci., 2002, 1, 745–748 CAS.
- B. Paul, P. Rajaputra and Y. You, Photochem. Photobiol., 2011, 87, 1468–1473 CrossRef CAS.
- H. von Tappeiner and A. Jesionek, MMW Munch. Med. Wochenschr., 1903, 47, 2042–2044 Search PubMed.
- H. v. Tappeiner, Ergeb. Physiol., 1909, 8, 698–741 CrossRef.
- A. Policard, C. R. Seances Soc. Biol., 1924, 91, 1423–1424 Search PubMed.
- H. Auler and G. Banzer, Z. Krebsforsch., 1942, 53, 65–68 CrossRef CAS.
- R. L. Lipson and E. J. Baldes, Arch. Dermatol., 1960, 82, 508–516 CAS.
- R. L. Lipson, E. J. Baldes and A. M. Olsen, J. Natl. Cancer Inst., 1961, 26, 1–11 CAS.
- R. L. Lipson, E. J. Baldes and A. M. Olsen, J. Thorac. Cardiovasc. Surg., 1961, 42, 623–629 CAS.
- R. L. Lipson, E. J. Baldes and M. J. Gray, Cancer, 1967, 20, 2255–2257 CrossRef CAS.
- R. L. Lipson, E. J. Baldes and A. M. Olsen, Chest, 1964, 46, 676–679 CrossRef CAS.
- T. J. Dougherty, G. B. Grindey, R. Fiel, K. R. Weishaupt and D. G. Boyle, J. Natl. Cancer Inst., 1975, 55, 115–121 CAS.
- J. F. Kelly, Proc. R. Soc. Med., 1975, 68, 527–528 CAS.
- J. F. Kelly, M. E. Snell and M. C. Berenbaum, Br. J. Cancer, 1975, 31, 237–244 CrossRef CAS.
- Y.-J. Hsieh, C.-C. Wu, C.-J. Chang and J.-S. Yu, J. Cell. Physiol., 2003, 194, 363–375 CrossRef CAS.
- M. Wainwright, Chem. Soc. Rev., 1996, 25, 351–359 RSC.
- D. E. J. G. J. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS.
- R. Bonnett, Chem. Soc. Rev., 1995, 24, 19–33 RSC.
- E. D. Sternberg, D. Dolphin and C. Bruckner, Tetrahedron, 1998, 54, 4151–4202 CrossRef CAS.
- R. R. Allison, G. H. Downie, R. Cuenca, X.-H. Hu, C. J. H. Childs and C. H. Sibata, Photodiagn. Photodyn. Ther., 2004, 1, 27–42 CrossRef CAS.
- E. S. Nyman and P. H. Hynninen, J. Photochem. Photobiol., B, 2004, 73, 1–28 CrossRef CAS.
- M. R. Detty, S. L. Gibson and S. J. Wagner, J. Med. Chem., 2004, 47, 3897–3915 CrossRef CAS.
- R. K. Pandey, L. N. Goswami, Y. Chen, A. Gryshuk, J. R. Missert, A. Oseroff and T. J. Dougherty, Lasers Surg. Med., 2006, 38, 445–467 CrossRef.
- A. E. O'Connor, W. M. Gallagher and A. T. Byrne, Photochem. Photobiol., 2009, 85, 1053–1074 CrossRef CAS.
- A. J. Bullous, C. M. Alonso and R. W. Boyle, Photochem. Photobiol. Sci., 2011, 10, 721–750 CAS.
- M. Ethirajan, Y. H. Chen, P. Joshi and R. K. Pandey, Chem. Soc. Rev., 2011, 40, 340–362 RSC.
- J. F. Lovell, T. W. Liu, J. Chen and G. Zheng, Chem. Rev., 2010, 110, 2839–2857 CrossRef CAS.
- S. Gross, A. Gilead, A. Scherz, M. Neeman and Y. Salomon, Nat. Med., 2003, 9, 1327–1331 CrossRef CAS.
- L. B. Josefsen and R. W. Boyle, Br. J. Pharmacol., 2008, 154, 1–3 CrossRef CAS.
- M. O. Senge, Photodiagn. Photodyn. Ther., 2012, 9, 170–179 CrossRef CAS.
- M. Mitsunaga, M. Ogawa, N. Kosaka, L. T. Rosenblum, P. L. Choyke and H. Kobayashi, Nat. Med., 2011, 17, 1685–1691 CrossRef CAS.
- J. P. Celli, B. Q. Spring, I. Rizvi, C. L. Evans, K. S. Samkoe, S. Verma, B. W. Pogue and T. Hasan, Chem. Rev., 2010, 110, 2795–2838 CrossRef CAS.
- S. S. Kelkar and T. M. Reineke, Bioconjugate Chem., 2011, 22, 1879–1903 CrossRef CAS.
- G. Zilidis, F. Aziz, S. Telara and M. S. Eljamel, Photodiagn. Photodyn. Ther., 2008, 5, 264–266 CrossRef CAS.
- M. S. Eljamel, Photodiagn. Photodyn. Ther., 2008, 5, 260–263 CrossRef CAS.
- M. Mitsunaga, T. Nakajima, K. Sano, P. L. Choyke and H. Kobayashi, Bioconjugate Chem., 2012, 23, 604–609 CrossRef.
- G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184–1201 CrossRef CAS.
- R. P. Haugland, The Handbook: A guide to Fluorerscent Probes and Labeling Technologies, 10th edn, Invitrogen Corp., Eugene, OR, 2005 Search PubMed.
- K. Umezawa, Y. Nakamura, H. Makino, D. Citterio and K. Suzuki, J. Am. Chem. Soc., 2008, 130, 1550 CrossRef CAS.
- M. A. T. Rogers, Nature, 1943, 151, 504 CrossRef CAS.
- M. A. T. Rogers, J. Chem. Soc., 1943, 590–596 RSC.
- A. Treibs and F. H. Kreuzer, Justus Liebigs Ann. Chem., 1986, 718, 208 Search PubMed.
- J. Killoran, L. Allen, J. F. Gallagher, W. M. Gallagher and D. F. O'Shea, Chem. Commun., 2002, 1862–1863 RSC.
- T. Yogo, Y. Urano, Y. Ishitsuka, F. Maniwa and T. Nagano, J. Am. Chem. Soc., 2005, 127, 12162–12163 CrossRef CAS.
- A. T. Byrne, A. E. O'Connor, M. Hall, J. Murtagh, K. O'Neill, K. M. Curran, K. Mongrain, J. A. Rousseau, R. Lecomte, S. McGee, J. J. Callanan, D. F. O'Shea and W. M. Gallagher, Br. J. Cancer, 2009, 101, 1565–1573 CrossRef CAS.
- IUPAC Compendium of Chemical Terminology, ed. A. D. McNaught and A. Wilkinson, Blackwell Scientific Publications, Oxford, UK, 2nd edn, 1997, vol. 68, p. 2245 Search PubMed.
- E. Deniz, G. C. Isbasar, O. A. Bozdemir, L. T. Yildirim, A. Siemiarczuk and E. U. Akkaya, Org. Lett., 2008, 10, 3401–3403 CrossRef CAS.
- Z. Dost, S. Atilgan and E. U. Akkaya, Tetrahedron, 2006, 62, 8484–8488 CrossRef CAS.
- A. Cihaner and F. Algi, React. Funct. Polym., 2009, 69, 62–67 CrossRef CAS.
- Y. Hayashi, S. Yamaguchi, W. Y. Cha, D. Kim and H. Shinokubo, Org. Lett., 2011, 13, 2992–2995 CrossRef CAS.
- M. T. K. Shah, M.-L. Soong, L. T. Wolford, J. H. Boyer, I. R. Politzer and T.G. Pavlopoulos, Heteroat. Chem., 1990, 1, 389 CrossRef CAS.
- A. Gorman, J. Killoran, C. O'Shea, T. Kenna, W. M. Gallagher and D. F. O'Shea, J. Am. Chem. Soc., 2004, 126, 10619–10631 CrossRef CAS.
- P. Batat, M. Cantuel, G. Jonusauskas, L. Scarpantonio, A. Palma, D. F. O'Shea and N. D. McClenaghan, J. Phys. Chem. A, 2011, 115, 14034–14039 CrossRef CAS.
- L. Jiao, W. Pang, J. Zhou, Y. Wei, X. Mu, G. Bai and E. Hao, J. Org. Chem., 2011, 76, 9988–9996 CrossRef CAS.
- X. Li, S. Huang and Y. Hu, Org. Biomol. Chem., 2012, 10, 2369–2372 CAS.
- S. G. Awuah, J. Polreis, V. Biradar and Y. You, Org. Lett., 2011 Search PubMed.
- M. J. Ortiz, A. R. Agarrabeitia, G. Duran-Sampedro, J. Bañuelos Prieto, T. A. Lopez, W. A. Massad, H. A. Montejano, N. A. García and I. L. Arbeloa, Tetrahedron, 2012, 68, 1153–1162 CrossRef CAS.
- N. Adarsh, R. R. Avirah and D. Ramaiah, Org. Lett., 2010, 12, 5720–5723 CrossRef CAS.
- N. Adarsh, R. R. Avirah and D. Ramaiah, Org. Lett., 2011, 13, 2146–2146 CrossRef CAS.
- S. H. Lim, C. Thivierge, P. Nowak-Sliwinska, J. Han, H. van den Bergh, G. Wagnieres, K. Burgess and H. B. Lee, J. Med. Chem., 2010, 53, 2865–2874 CrossRef CAS.
- D. Wang, J. Fan, X. Gao, B. Wang, S. Sun and X. Peng, J. Org. Chem., 2009, 74, 7675–7683 CrossRef CAS.
- Z. Li, E. Mintzer and R. Bittman, J. Org. Chem., 2006, 71, 1718–1721 CrossRef CAS.
- V. Gottfried, E. S. Lindenbaum and S. Kimel, Int. J. Radiat. Biol., 1991, 60, 349–354 CrossRef CAS.
- V. Gottfried, E. S. Lindenbaum and S. Kimel, J. Photochem. Photobiol., B, 1992, 12, 204–207 CrossRef CAS.
- R. Weissleder, Nat. Biotechnol., 2001, 19, 316–317 CrossRef CAS.
- J. Chen, A. Burghart, A. Derecskei-Kovacs and K. Burgess, J. Org. Chem., 2000, 65, 2900–2906 CrossRef CAS.
- Y. J. Mei, P. A. Bentley and W. Wang, Tetrahedron Lett., 2006, 47, 2447–2449 CrossRef CAS.
- C. Thivierge, R. Bandichhor and K. Burgess, Org. Lett., 2007, 9, 2135–2138 CrossRef CAS.
- T. Rohand, W. W. Qin, N. Boens and W. Dehaen, Eur. J. Org. Chem., 2006, 4658–4663 CrossRef CAS.
- L. J. Jiao, C. J. Yu, T. Uppal, M. M. Liu, Y. Li, Y. Y. Zhou, E. H. Hao, X. K. Hu and M. G. H. Vicente, Org. Biomol. Chem., 2010, 8, 2517–2519 CAS.
- J. Y. Han, O. Gonzalez, A. Aguilar-Aguilar, E. Pena-Cabrera and K. Burgess, Org. Biomol. Chem., 2009, 7, 34–36 CAS.
- T. Rohand, M. Baruah, W. W. Qin, N. Boens and W. Dehaen, Chem. Commun., 2006, 266–268 RSC.
- T. Rousseau, A. Cravino, T. Bura, G. Ulrich, R. Ziessel and J. Roncali, Chem. Commun., 2009, 1673–1675 RSC.
- J. Y. Liu, E. A. Ermilov, B. Roder and D. K. P. Ng, Chem. Commun., 2009, 1517–1519 RSC.
- O. Buyukcakir, O. A. Bozdemir, S. Kolemen, S. Erbas and E. U. Akkaya, Org. Lett., 2009, 11, 4644–4647 CrossRef CAS.
- M. Wada, S. Ito, H. Uno, T. Murashima, N. Ono, T. Urano and Y. Urano, Tetrahedron Lett., 2001, 42, 6711–6713 CrossRef CAS.
- Z. Shen, H. Rohr, K. Rurack, H. Uno, M. Spieles, B. Schulz, G. Reck and N. Ono, Chem.–Eur. J., 2004, 10, 4853–4871 CrossRef CAS.
- S. Goeb and R. Ziessel, Org. Lett., 2007, 9, 737–740 CrossRef CAS.
- G. Ulrich, S. Goeb, A. De Nicola, P. Retailleau and R. Ziessel, Synlett, 2007, 1517–1520 CrossRef CAS.
- L. J. Jiao, C. J. Yu, M. M. Liu, Y. C. Wu, K. B. Cong, T. Meng, Y. Q. Wang and E. H. Hao, J. Org. Chem., 2010, 75, 6035–6038 CrossRef CAS.
- J. Griffiths, in Modern Colorants: synthesis and structure, Blackie Academic and Professional, Glasgow, U. K., 1995 Search PubMed.
- G. Qian and Z. Y. Wang, Chem.–Asian J., 2010, 5, 1006–1029 CrossRef CAS.
- J. Chen, A. Burghart, A. Derecskei-Kovacs and K. Burgess, J. Org. Chem., 2000, 65, 2900–2906 CrossRef CAS.
- A. Coskun, E. Deniz and E. U. Akkaya, Org. Lett., 2005, 7, 5187–5189 CrossRef CAS.
- S. Atilgan, Z. Ekmekci, A. L. Dogan, D. Guc and E. U. Akkaya, Chem. Commun., 2006, 4398–4400 RSC.
- H. He, P. C. Lo, S. L. Yeung, W. P. Fong and D. K. Ng, J. Med. Chem., 2011, 54, 3097–3102 CrossRef CAS.
- M. R. Hamblin, J. L. Miller, I. Rizvi, B. Ortel, E. V. Maytin and T. Hasan, Cancer Res., 2001, 61, 7155–7162 CAS.
- S. K. Sahoo, T. Sawa, J. Fang, S. Tanaka, Y. Miyamoto, T. Akaike and H. Maeda, Bioconjugate Chem., 2002, 13, 1031–1038 CrossRef CAS.
- V. Rapozzi, M. Zacchigna, S. Biffi, C. Garrovo, F. Cateni, M. Stebel, S. Zorzet, G. M. Bonora, S. Drioli and L. Xodo, Cancer Biol. Ther., 2010, 10, 471–482 CrossRef CAS.
- H. He, P.-C. Lo, S.-L. Yeung, W.-P. Fong and D. K. P. Ng, Chem. Commun., 2011, 47, 4748–4750 RSC.
- S. Erbas, A. Gorgulu, M. Kocakusakogullari and E. U. Akkaya, Chem. Commun., 2009, 4956–4958 RSC.
- S. Kim, T. Y. Ohulchanskyy, A. Baev and P. N. Prasad, J. Mater. Chem., 2009, 19, 3181–3188 RSC.
- S. K. Lower and M. A. Elsayed, Chem. Rev., 1996, 66, 199 CrossRef.
- A. K. Chandra, N. J. Turro, A. L. Lyons and P. Stone, J. Am. Chem. Soc., 1978, 100, 4964–4968 CrossRef CAS.
- J. C. Koziar and D. O. Cowan, Acc. Chem. Res., 1978, 11, 334–341 CrossRef CAS.
- S. P. Mcglynn, R. Sunseri and N. Christod, J. Chem. Phys., 1962, 37, 1818 CrossRef CAS.
- S. P. Mcglynn, G. W. Daigre, N. Christodoyleas and M. J. Reynolds, J. Phys. Chem., 1962, 66, 2499 CrossRef CAS.
- P. Yuster and S. I. Weissman, J. Chem. Phys., 1949, 17, 1182–1188 CrossRef CAS.
- D. S. Mcclure, J. Chem. Phys., 1949, 17, 665–666 CrossRef CAS.
- D. S. Mcclure, J. Chem. Phys., 1949, 17, 905–913 CrossRef CAS.
- W. M. Gallagher, L. T. Allen, C. O'Shea, T. Kenna, M. Hall, A. Gorman, J. Killoran and D. F. O'Shea, Br. J. Cancer, 2005, 92, 1702–1710 CrossRef CAS.
- B. Chen, B. W. Pogue, P. J. Hoopes and T. Hasan, Crit. Rev. Eukaryotic Gene Expression, 2006, 16, 279–305 CrossRef CAS.
- B. Chen, B. W. Pogue, J. M. Luna, R. L. Hardman, P. J. Hoopes and T. Hasan, Clin. Cancer Res., 2006, 12, 917–923 CrossRef CAS.
- A. E. O'Connor, M. M. Mc Gee, Y. Likar, V. Ponomarev, J. J. Callanan, F. D. O'Shea, A. T. Byrne and W. M. Gallagher, Int. J. Cancer, 2012, 130, 705–715 CrossRef CAS.
- T. Maisch, Mini-Rev. Med. Chem., 2009, 9, 974–983 CAS.
- G. Bertoloni, F. Rossi, G. Valduga, G. Jori and J. Vanlier, FEMS Microbiol. Lett., 1990, 71, 149–155 CrossRef CAS.
- Y. Nitzan, M. Gutterman, Z. Malik and B. Ehrenberg, Photochem. Photobiol., 1992, 55, 89–96 CrossRef CAS.
- Z. Malik, H. Ladan and Y. Nitzan, J. Photochem. Photobiol., B, 1992, 14, 262–266 CrossRef CAS.
- M. R. Hamblin, D. A. O'Donnell, N. Murthy, C. H. Contag and T. Hasan, Photochem. Photobiol., 2002, 75, 51–57 CrossRef CAS.
- T. Dai, G. P. Tegos, T. Zhiyentayev, E. Mylonakis and M. R. Hamblin, Lasers Surg. Med., 2010, 42, 38–44 CrossRef.
- R. Dosselli, M. Gobbo, E. Bolognini, S. Campestrini and E. Reddi, ACS Med. Chem. Lett., 2010, 1, 35–38 CrossRef CAS.
- T. Maisch, C. Bosl, R. M. Szeimies, N. Lehn and C. Abels, Antimicrob. Agents Chemother., 2005, 49, 1542–1552 CrossRef CAS.
- H. Mohr, B. Lambrecht and A. Selz, Immunol. Invest., 1995, 24, 73–85 CrossRef CAS.
- M. Wainwright, H. Mohr and W. H. Walker, J. Photochem. Photobiol., B, 2007, 86, 45–58 CrossRef CAS.
- O. E. Akilov, S. Kosaka, K. O'Riordan, X. Z. Song, M. Sherwood, T. J. Flotte, J. W. Foley and T. Hasan, Chem. Biol., 2006, 13, 839–847 CrossRef CAS.
- D. O. Frimannsson, M. Grossi, J. Murtagh, F. Paradisi and D. F. O'Shea, J. Med. Chem., 2010, 53, 7337–7343 CrossRef CAS.
- Y. Cakmak, S. Kolemen, S. Duman, Y. Dede, Y. Dolen, B. Kilic, Z. Kostereli, L. T. Yildirim, A. L. Dogan, D. Guc and E. U. Akkaya, Angew. Chem., Int. Ed., 2011, 50, 11937–11941 CrossRef CAS.
- W. Pang, X.-F. Zhang, J. Zhou, C. Yu, E. Hao and L. Jiao, Chem. Commun., 2012, 48, 5437–5439 RSC.
- A. N. Amin, M. E. El-Khouly, N. K. Subbaiyan, M. E. Zandler, S. Fukuzumi and F. D'Souza, Chem. Commun., 2012, 48, 206–208 RSC.
- L. Huang, X. Yu, W. Wu and J. Zhao, Org. Lett., 2012, 14, 2594–2597 CrossRef CAS.
- S. Hoogendoorn, A. E. Blom, L. I. Willems, G. A. van der Marel and H. S. Overkleeft, Org. Lett., 2011, 13, 5656–5659 CrossRef CAS.
- S. Hoogendoorn, K. L. Habets, S. Passemard, J. Kuiper, G. A. van der Marel, B. I. Florea and H. S. Overkleeft, Chem. Commun., 2011, 47, 9363–9365 RSC.
- Y. Urano, D. Asanuma, Y. Hama, Y. Koyama, T. Barrett, M. Kamiya, T. Nagano, T. Watanabe, A. Hasegawa, P. L. Choyke and H. Kobayashi, Nat. Med., 2009, 15, 104–109 CrossRef CAS.
- T. Miura, Y. Urano, K. Tanaka, T. Nagano, K. Ohkubo and S. Fukuzumi, J. Am. Chem. Soc., 2003, 125, 8666–8671 CrossRef CAS.
- T. Yogo, Y. Urano, A. Mizushima, H. Sunahara, T. Inoue, K. Hirose, M. Iino, K. Kikuchi and T. Nagano, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 28–32 CrossRef CAS.
- S. O. McDonnell, M. J. Hall, L. T. Allen, A. Byrne, W. M. Gallagher and D. F. O'Shea, J. Am. Chem. Soc., 2005, 127, 16360–16361 CrossRef CAS.
- M. N. Win and C. D. Smolke, Science, 2008, 322, 456–460 CrossRef CAS.
- A. P. de Silva, M. R. James, B. O. McKinney, D. A. Pears and S. M. Weir, Nat. Mater., 2006, 5, 787–790 CrossRef.
- R. J. Amir, M. Popkov, R. A. Lerner, C. F. Barbas 3rd and D. Shabat, Angew. Chem., Int. Ed., 2005, 44, 4378–4381 CrossRef CAS.
- P. A. de Silva, N. H. Q. Gunaratne and C. P. McCoy, Nature, 1993, 364, 42–44 CrossRef.
- K. Szacilowski, Chem. Rev., 2008, 108, 3481–3548 CrossRef CAS.
- P. Montcourrier, P. H. Mangeat, C. Valembois, G. Salazar, A. Sahuquet, C. Duperray and H. Rochefort, J. Cell. Sci., 1994, 107, 2381–2391 Search PubMed.
- I. L. Cameron, N. K. R. Smith, T. B. Pool and R. L. Sparks, Cancer Res., 1980, 40, 1493–1500 CAS.
- S. Ozlem and E. U. Akkaya, J. Am. Chem. Soc., 2008, 131, 48–49 CrossRef.
- A. D. Quartarolo, N. Russo and E. Sicilia, Chem.–Eur. J., 2006, 12, 6797–6803 CrossRef CAS.
- S. Duman, Y. Cakmak, S. Kolemen, E. U. Akkaya and Y. Dede, J. Org. Chem., 2012, 77, 4516–4527 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: a summary table of BODIPY-based photosensitizers: structures, optical properties, light dosimetry, cell line and drug doses. See DOI: 10.1039/c2ra21404k |
| This journal is © The Royal Society of Chemistry 2012 |