Oxidative rearrangement of indoles to oxindoles†
Wesley J.
Moran
*,
Kerry L.
MacRory
and
Arantxa
Rodríguez
*
Department of Chemical & Biological Sciences, University of Huddersfield, Queensgate, Huddersfield HD1 3DH, UK. E-mail: w.j.moran@hud.ac.uk; a.r.menendez@hud.ac.uk; Tel: +44 (0)1484 473741
First published on 16th August 2012
Abstract
An unexpected oxidative rearrangement was observed when indoles substituted with 2-hydroxymalonates were subjected to typical iodination conditions. This unusual and unprecedented transformation is successful for a number of derivatives.
Indoles are important structural motifs present in many compounds of pharmacological significance.1–3 Methods to elaborate the structure of simple indoles into more substituted variants are of immense importance to enable the synthesis of new structures of potential value.4
During an attempted total synthesis of a bisindole natural product we required the introduction of an iodine atom at the two-position of a 1,3-disubstituted indole. Due to a lack of literature methods for the iodination of 1,3-disubstituted indoles,5 we decided to treat indole derivative 1a with silver trifluoroacetate and iodine in THF at room temperature. However, instead of the desired iodination, an oxidative rearrangement occurred generating the oxindole 2a (Scheme 1).
 | ||
| Scheme 1 Attempted iodination of substituted indole 1. | ||
The oxidative rearrangement of tertiary allylic alcohols to α,β-unsaturated ketones has been known for some time (Scheme 2a),6 however the generation of α,β-unsaturated amides by this route is unknown. Indoles are known to undergo oxidative rearrangement to oxindoles upon treatment with hypochlorite or other oxidant species (Scheme 2b),7 but this is distinct from the work reported herein.
 | ||
| Scheme 2 Examples of rearrangements in the literature. | ||
At this stage, we looked to optimize the reaction conditions for this oxidative rearrangement (Table 1). Initially different silver salts were tested, but no rearrangement occurred with any but the silver trifluoroacetate (entries 1–4). Running the reaction without either the iodine or the silver salt led to no conversion (entries 5–6). Interestingly, running the reaction with one equivalent of N-iodosuccinimide did lead to product formation, albeit in low yield (entry 7). Increasing the amount of N-iodosuccinimide to five equivalents did increase conversion to product however a low yield was still obtained (entry 8). In addition, no desired reaction was observed using typical conditions for the oxidative rearrangement of indoles as described above, i.e. with tert-butylhypochlorite as oxidant in dichloromethane (entry 9).
| Entry | Conditionsa | Yield/%b |
|---|---|---|
| a All reactions run at room temperature. b Yield of pure isolated product 2a. | ||
| 1 | 1.2 equiv AgO2CCF3, 1 equiv I2, THF | 61 |
| 2 | 1.2 equiv AgOAc, 1 equiv I2, THF | 0 |
| 3 | 1.2 equiv AgBF4, 1 equiv I2, THF | 0 |
| 4 | 1.2 equiv AgNO3, 1 equiv I2, THF | 0 |
| 5 | 1.2 equiv AgO2CCF3, THF | 0 |
| 6 | 1 equiv I2, THF | 0 |
| 7 | 1 equiv N-iodosuccinimide, THF | 21 |
| 8 | 5 equiv N-iodosuccinimide, THF | 30 |
| 9 | t-BuOCl, CH2Cl2 | 0 |
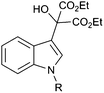
We then prepared a range of hydroxymalonate substituted indoles simply by heating to reflux toluene solutions of indoles with commercially available diethyl ketomalonate (Table 2).8 This reaction worked very well for 1-methyl indole 3a and 1-ethyl indole 3b, however 1-benzyl 3c and 1-phenyl 3d indoles only reacted in moderate yields (entries 1–4). A range of substituted 1-methyl indoles were subjected to these conditions, however only indoles with bromo and chloro substituents were found to be successful (entries 5–7). Reactions of indoles with methoxy, ethyl, methyl and nitro substituents gave very messy reaction mixtures and no desired product could be isolated.

In order to prepare indole substrates with electron donating substituents, alternative reaction conditions were required. A range of Lewis acids were screened and it was found that cerium chloride efficiently promoted the reaction at room temperature in dichloromethane.9 Indole 3h bearing a 5-methoxy substituent underwent smooth addition providing the product in 95% yield (entry 8). Indoles with a 7-ethyl and a 5-methyl substituent provided the products in 74% and 69% respectively (entries 9 and 10).
With these substituted indoles in hand, the rearrangement reactions were investigated (Table 3).10 1-Ethyl indole 1b rearranged in a moderate 42% yield (entry 2), whereas the 1-benzyl indole 1c provided a 62% yield (entry 3). The 1-phenyl indole 1d was not as effective in the rearrangement, furnishing only 34% yield of product (entry 4), showing that methyl and benzyl activating groups are preferable. The remainder of the mass balance was unidentifiable in all of these reactions. The bromo and chloro substituted indoles 1e–g rearranged in moderate yields, 51%, 45% and 34% respectively (entries 5–7), however unreacted starting material could be isolated from these reaction mixtures and resubjected to the reaction conditions.11 The indoles with electron donating substituents (1h–j) were reacted at 0 °C as superior yields were obtained compared to at room temperature. In these cases, complete conversion of the starting material was evident with the identity of the rest of the mass balance being unknown. Oxindole 2h was isolated in 68% yield, which demonstrates that electron donating substituents aid this transformation (entry 8). The presence of alkyl substituents led to rearrangement to oxindoles 2i and 2j in 58% and 53% respectively (entries 9 and 10).
| Entry | Indole | Product | Yield/%b |
|---|---|---|---|
| a 1.2 equiv AgO2CCF3, 1 equiv I2, THF, rt, 16 h. b Yield of pure isolated product. c Remainder of mass balance is unreacted starting material. d Reaction run at 0 °C. | |||
|

|
||
| 1 | R = Me (1a) | 61 (2a) | |
| 2 | R = Et (1b) | 42 (2b) | |
| 3 | R = Bn (1c) | 62 (2c) | |
| 4 | R = Ph (1d) | 34 (2d) | |
| 5 |
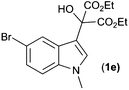 (1e) (1e) |

|
51c (2e) |
| 6 |
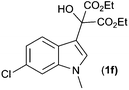 (1f) (1f) |

|
45c (2f) |
| 7 |
 (1g) (1g) |
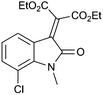
|
34c (2g) |
| 8 |
 (1h) (1h) |
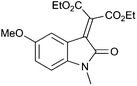
|
68d (2h) |
| 9 |
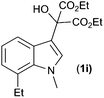 (1i) (1i) |
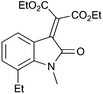
|
58d (2i) |
| 10 |
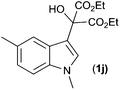 (1j) (1j) |

|
53d (2j) |
An X-ray crystal structure of the N-ethyl oxindole 2b was obtained to provide confirmation of the structure (Fig. 1 and ESI†). Interestingly, this shows that only one of the two ester groups is in conjugation with the alkene in the solid state.
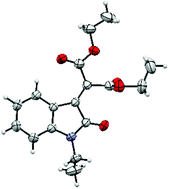 | ||
| Fig. 1 X-ray crystal structure of N-ethyl oxindole 2b. | ||
In an attempt to elucidate the mechanism of this process a number of experiments were undertaken; however, a lack of availability of suitably 18O-labelled compounds hampered the progress. The possibility of adventitious water being key to rearrangement in the reactions mediated by either N-iodosuccinimide or silver trifluoroacetate was explored. In both cases, addition of five equivalents of water led to considerably messier reaction mixtures however the yield was slightly augmented in the former case and diminished in the latter. Repeating both reactions with H218O led to similar results, however 18O was not incorporated into either product. In addition, running the reactions with activated 4 Å molecular sieves did not lead to product inhibition. It remains unclear as to whether the carbonyl oxygen in the product originates from the hydroxyl of the starting material or from the N-iodosuccinimide or silver trifluoroacetate. Attempts to make 18O-labelled diethylketomalonate were unsuccessful.
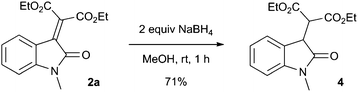
These oxindole products are amenable to further synthetic transformations such as the chemoselective reduction of the alkene with sodium borohydride (eqn (1)).
 | (1) |
In conclusion, we have demonstrated an unprecedented oxidative rearrangement of hydroxymalonate substituted indoles to oxindoles.
We thank Dr Craig Rice for acquiring the X-ray crystal structure, and the University of Huddersfield for funding.
References
- C. Sánchez, C. Méndez and J. A. Salas, Nat. Prod. Rep., 2006, 23, 1007 RSC.
- T. Kawasaki and K. Higuchi, Nat. Prod. Rep., 2005, 22, 761 RSC.
- R. J. Sundberg, Indoles; Academic Press: London, U.K., 1996 Search PubMed.
- For selected recent examples, see: (a) S. Lucarini, F. Bartoccini, F. Battistoni, G. Diamantini, G. Piersanti, M. Righi and G. Spadoni, Org. Lett., 2010, 12, 3844 CrossRef CAS; (b) Z. Shi, Y. Cui and N. Jiao, Org. Lett., 2010, 12, 2908 CrossRef CAS; (c) C. C. Silveira, S. R. Mendes, L. Wolf and G. M. Martins, Tetrahedron Lett., 2010, 51, 4560 CrossRef CAS; (d) M. Bandini and A. Eichholzer, Angew. Chem., Int. Ed., 2009, 48, 9608 CrossRef CAS; (e) M. Yamashita, H. Horiguchi, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2009, 74, 7481 CrossRef CAS; (f) G. R. Humphrey and J. T. Kuethe, Chem. Rev., 2006, 106, 2875 CrossRef CAS.
- For the bromination and chlorination of 1, 3-disubstituted indoles, see: S. Tang, J.-H. Li, Y.-X. Xie and N.-X. Wang, Synthesis, 2007, 1535 CAS.
- (a) J.-M. Vatèle, Tetrahedron, 2010, 66, 904 CrossRef; (b) For other recent examples, see: M. Uyanik, R. Fukatsu and K. Ishihara, Org. Lett., 2009, 11, 3470 CrossRef CAS; (c) M. Shibuya, S. Ito, M. Takahashi and Y. Iwabuchi, Org. Lett., 2008, 10, 4715 CrossRef CAS; (d) M. Shibuya, S. Ito, M. Takahashi and Y. Iwabuchi, Org. Lett., 2004, 6, 4303 CrossRef CAS.
- (a) C. Poriel, M. Lachia, C. Wilson, J. R. Davies and C. J. Moody, J. Org. Chem., 2007, 72, 2978 CrossRef CAS; (b) For other examples, see: T. Lindel, L. Bräuchle, G. Golz and P. Böhrer, Org. Lett., 2007, 9, 283 CrossRef CAS; (c) M. Lachia, C. Poriel, A. M. Z. Slawin and C. J. Moody, Chem. Commun., 2007, 286 RSC; (d) M. Pettersson, D. Knueppel and S. F. Martin, Org. Lett., 2007, 9, 4623 CrossRef CAS; (e) P. S. Baran and J. M. Richter, J. Am. Chem. Soc., 2005, 127, 15394 CrossRef CAS; (f) M. Ito, C. W. Clark, M. Mortimore, J. B. Goh and S. F. Martin, J. Am. Chem. Soc., 2001, 123, 8003 CrossRef CAS; (g) X. Zhang and C. S. Foote, J. Am. Chem. Soc., 1993, 115, 8867 CrossRef CAS; (h) D. V. C. Awang, A. Vincent and D. Kindack, Can. J. Chem., 1984, 62, 2667 CrossRef CAS; (i) A. Walser, J. F. Blount and R. I. Fryer, J. Org. Chem., 1973, 38, 3077 CrossRef CAS; (j) R. M. Acheson, R. W. Snaith and J. M. Vernon, J. Chem. Soc., 1964, 3229 RSC; (k) N. Finch and W. I. Taylor, J. Am. Chem. Soc., 1962, 84, 3871 CrossRef CAS.
- 1-Methylindole and some 1-methyl-2-vinylindoles are known to react with diethyl ketomalonate: (a) U. Pindur and M.-H. Kim, Tetrahedron, 1989, 45, 6427 CrossRef CAS; (b) U. Pindur and M.-H. Kim, Arch. Pharm., 1992, 325, 353 CrossRef CAS.
- Other Lewis acids have been reported to promote double addition of indoles to diethyl ketomalonate or to mediate indole dimerization: see Ref. 6a.
- Typical experimental procedure: Iodine (83 mg, 0.328 mmol) in dry THF (0.90 mL) was added via cannula to silver trifluoroacetate (87 mg, 0.393 mmol) and diethyl 2-hydroxy-2-(1-methyl-1H-indol-3-yl)malonate 1a (100 mg, 0.328 mmol) in dry THF (1.8 mL). The reaction mixure was stirred at room temperature for 16 h under a nitrogen atmosphere and then quenched with saturated sodium thiosulfate and extracted with EtOAc. The organic layer was dried over MgSO4, concentrated and purified by flash chromatography on silica gel (5
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 petroleum ether/ethyl acetate) to provide diethyl 2-(1-methyl-2-oxoindolin-3-ylidene)malonate 2a as a red solid (61 mg, 61%).
:1 petroleum ether/ethyl acetate) to provide diethyl 2-(1-methyl-2-oxoindolin-3-ylidene)malonate 2a as a red solid (61 mg, 61%). - Running the reactions with more equivalents of silver salt and iodine did not lead to noticeable increases in yield.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC reference number 828908. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra21800c |
| This journal is © The Royal Society of Chemistry 2012 |