New phospha-palladacycles: efficient catalysts in the formylation of aryl chlorides†
J. Luis Serrano*a, José Péreza, Luis Garcíaa, Gregorio Sánchezb, Joaquín Garcíab, Kriti Tyagic and Anant Kapdi*c
aDepartamento de Ingeniería Minera, Geológica y Cartográfica. Universidad Politécnica de Cartagena, Área de Química Inorgánica, 30203 Cartagena, Spain
bDepartamento de Química Inorgánica, Universidad de Murcia, 30071 Murcia, Spain
cInstitute of Chemical Technology, Mumbai Nathalal Road, Matunga, Mumbai-400019, India
First published on 3rd October 2012
Abstract
The direct reaction between [{Pd(μ-AcO)(CˆP)}2] 1 and cyclic imides to yield new dinuclear cyclometallated palladium(II) complexes containing –NCO– bridging imidates of general formula [{Pd(μ-NCO)(CˆP)}2] (CˆP = CH2C6H4P(o-tolyl)2; –NCO– = succinimidate (suc) 2, maleimidate (mal) 3, phthalimidate (phthal) 4 or saccharinate (sacc) 5 is described. Spectroscopic techniques and structural characterisation by X-ray diffraction of complex 2 confirmed the proposed formulae. The new dimeric complexes are shown to be excellent catalysts for the selective formylation of aryl halides under CO-free conditions to obtain synthetically important aromatic aldehydes. Yields are diminished by steric hindrance around the aryl halides.
Introduction
Since the first descriptions of cyclopalladation in the 1960s1 to date several reviews have been dedicated to those different aspects of chemistry of palladacycles that have brought them to a prominent place in organometallic palladium chemistry. Thus, their synthesis and structural characterization, their applications in organic synthesis or organometallic catalysis, and their presence in medicinal and biological chemistry or use as chiral auxiliaries, mesogenic and luminescent agents have been widely explored.2 Probably the most striking application comes from their use as catalysts in C–C and C–heteroatom bond forming reactions, a field in which palladacycles changed from being perceived as dead-end deactivation products to be considered among the most useful reagents, and therefore the object of considerable attention since then.3 Complexes containing a cyclopalladated (di-o-tolylphoshino)benzyl fragment such as 1 illustrate like no others this revaluation experienced after the original articles of Herrmann, Beller and co-workers,4 that has kept since 1995 a permanent interest in phosphapalladacycles mainly focused in the design of new CˆP systems.2,3,5 However, in contrast with complexes that contain CˆN cyclopalladated backbones whose chemistry has been widely expanded from the dinuclear acetate or chloride bridged precursors by reacting them with neutral or anionic ligands to yield di- or mononuclear derivatives, that in some cases improved the catalytic performance of the starting materials,3b only a few reports exploiting the reactivity of acetate bridges in 1 can be found.3a,4c,6In this sense, during the last few years we have taken advantage of binuclear acetate/hydroxo complexes of palladium, some of them with a cyclometalated backbone,7 in the preparation of a wide range of new compounds by means of a simple acid–base reaction without addition of other basic reagents.8 Regarding the application in catalysis of these new derivatives, we particularly succeeded with those containing imidate –CNO– bridges such as reported here. Thus, binuclear imidate cyclometalated palladium(II) complexes and mononuclear phosphine adducts have been tested in Stille,8a Suzuki and Sonogashira cross-couplings,7a,9 and phosphine-free anionic dinuclear imidate complexes with a palladacyclopentadiene backbone have displayed impressive performances in Stille couplings.10 Despite the potential of imidato complexes not only in catalysis, but also in medicinal11 or supramolecular chemistry,12 their coordination chemistry has not been much explored, with saccharinate complexes being most fully characterized, mainly by Yilmaz and co-workers.13
On the other hand, aromatic aldehydes have found a prominent place in synthesis due to the highly reactive nature of the aldehyde functional group. These are readily transformed into different functional groups that can provide a handle towards achieving the synthesis of complex biologically active molecules. Given the importance of aromatic aldehydes over the years, several novel protocols have been developed that have tried to replace the more traditional approaches14 which have suffered from the formation of undesired side products and waste. Several formylating reagents have been employed such as formate salts, carbon monoxide under reductive formylation conditions15,16 as well as the environmentally benign formylation source CO/H2 (synthesis gas).17 Although in all these cases high reactivity could be achieved, the use of relatively hazardous chemical reagents or gases at elevated temperatures and pressures are far from ideal.
Like in the usual C–C, C–O and C–N bond-forming technologies,18 aryl chlorides are among the most desirable substrates due to their lower costs. However, the reduced reactivity associated with aryl chlorides because of their inherently high C–Cl bond strength makes these substrates synthetically more challenging towards different reactions.19,5a To the best of our knowledge, efficient palladium-catalyzed formylation of aryl chlorides can be accomplished via hydroformylation using either electron-rich and bulky phosphines or highly elaborate palladium complexes.15 In this paper we report the synthesis and characterization of new dinuclear imidate complexes that contain the cyclometalated (di-o-tolylphoshino)benzyl fragment by means of one-step reaction with the well known di-μ-acetate precursor. We also present here a mild and efficient CO-free protocol for the synthesis of aromatic aldehydes from aryl chlorides using these novel palladacyclic complexes, which display excellent selectivity of formylation vs. cyanation.
Results and discussion
Synthesis and characterization
In 1999 Herrmann and co-workers published the first review after the revival experienced by phosphapalladacycles a few years before.3a Although, mainly focused in catalytic applications, this article also collected the previous reports on CˆP-palladacycles20 and interestingly it gathered together most of the chemistry done on the acetate groups of the complex trans-di(μ-acetato)-bis[o-(di-o-tolylphosphino)benzyl]dipalladium(II) 1 and related complexes to date. Thus anion interchange with tetrabutylammonium halides produces poorly soluble di-μ-halide dimers4c and bridge-splitting with coordinating solvents or neutral donor ligands such as pyridine or PPh3 is reported to form soluble monomeric complexes,3a while acetylacetonate or 2-acylphenolate complexes are obtained by reacting 1 with 2,4-pentanodione (Hacac)6b,c or 2-acylphenoles, respectively.6a Whereas the presence of a strong base is needed for the success of this last reaction, direct treatment of 1 with Hacac produced the expected monomeric derivatives.This was found in our related studies and di-μ-acetate-complex 1 reacted with cyclic imides to give binuclear species of general formula [{Pd(μ-NCO)(CˆP)}2] (CˆP = CH2C6H4P(o-tolyl)2; –NCO– = imidate). The structures of the protonated imidate ligands with their abbreviations are shown in Scheme 1.
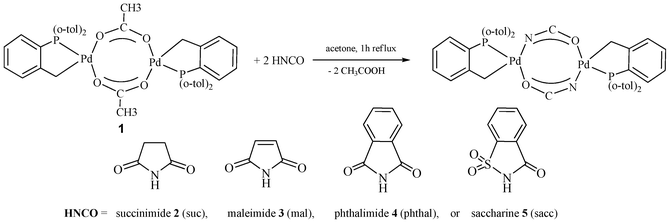 | ||
| Scheme 1 | ||
The specific conditions followed to prepare complexes 2–5 are given in the Experimental section. The new palladium complexes 2–5 are yellow air-stable solids and their IR spectra show absorptions assignable to the cyclometallated CˆP backbone around 500 cm−1,4c,21 with absence of relevant bands in the region 3000–3500 cm−1 and important variations in the carbonyl region indicating that acetate–imidate interchange has taken place. Two strong bands in the range 1728–1719 and 1632–1600 cm−1 appear now instead of the very strong band at 1578 cm−1 observed in 1. This suggests coordination of one imidate-carbonyl group to the metal as a part of an –NCO– bridging unit. According to previously reported data,22 a bridging structure would remove the two-fold symmetry of the free or N-bonded imidato ligand, increasing the intensity of the symmetric absorption. This higher frequency mode νsym(CO) is normally weak in cyclic imides and has negligible intensity compared with the antisymmetric ν(CO) absorption.
The dinuclearity of complexes 2–5 was confirmed by means of FAB mass spectrometry, as can be implied by the m/z values for the observed fragments. Spectra of the new bridged compounds show a similar fragmentation pattern which includes the peaks corresponding to M+ − imidate and M+/2. The abundances of the signals are consistent with the natural isotopic abundances.
Typical broad NMR resonances are observed, that in related compounds have been justified in terms of equilibria in solution caused by the interaction of the labile acetate bridges and deuterated solvents.3a,4c,6a In spite of this the spectra show the corresponding signals of the incoming imidate ligands, overlapped in the aromatic region with those of the cyclometallated backbone.
In the case of complex 2, its dinuclear nature has also been confirmed by single-crystal X-ray analysis. The corresponding ORTEP drawing is shown in Fig. 1 with relevant bond lengths and angles provided in the figure caption.
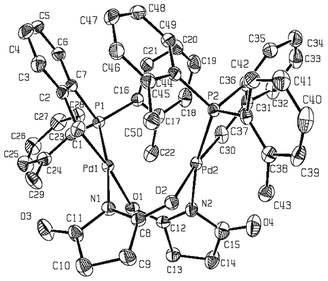 | ||
| Fig. 1 ORTEP diagram of complex 2 with atom numbering scheme; thermal ellipsoids are drawn at the 50% probability level. (CH3)2CO found in the unit cell is not shown. Selected bond lengths (Å) and angles (°): Pd(1)–C(1) 2.024(4), Pd(1)–N(1) 2.099(3), Pd(1)–O(1) 2.151(3), Pd(1)–P(1) 2.2330(10), Pd(2)–C(30) 2.028(4), Pd(2)–N(2) 2.101(3), Pd(2)–O(2) 2.158(3), Pd(2)–P(2) 2.2284(10), Pd(1)–Pd(2) 3.2338(4); C(1)–Pd(1)–N(1) 88.40(15), C(1)–Pd(1)–O(1) 172.92(15), C(1)–Pd(1)–P(1) 84.40(12), N(1)–Pd(1)–O(1) 87.58(13), N(1)–Pd(1)–P(1) 171.13(10), O(1)–Pd(1)–P(1) 99.03(8), C(30)–Pd(2)–N(2) 87.54(17), C(30)–Pd(2)–O(2) 173.34(16), C(30)–Pd(2)–P(2) 84.03(14), N(2)–Pd(2)–O(2) 88.37(13), N(2)–Pd(2)–P(2) 168.53(10), O(2)–Pd(2)–P(2) 99.27(9). | ||
The X-ray analysis has also established a head-to tail arrangement, the most common in this type of complexes. Regarding the cyclometallated CˆP ligands, an anti-arrangement of identical donor atoms is found. The structure displays a so-called open-book geometry accompanied by a Pd(1)–Pd(2) distance of 3.2338(4) Å. We have recently developed methods for the conformational classification of eight-membered rings.23 From this perspective the Pd(1)–N(1)–C(8)–O(2)–Pd(2)–N(2)–C(12)–O(1) ring displays a twist-boat conformation distorted by 38° (TB = 0.7719; BB = 0.2281 for σ = 10).The structure around the two palladium atoms may be described as nearly planar, and their deviation from the planar coordination has been quantified by measures of improper torsion angles: 4.10 and −3.94° for Pd(1); −3.74 and 5.77° for Pd(2) (both square pyramidal).24
Bond distances and angles for 2 are close to those displayed by related complexes containing a di-o-tolylphosphino)benzyl backbone that have been previously reported.25,3a,6a Following the classification of Dance and Scudder26 for PPh3 based on measures of torsion angles M–P–Cipso–C, the conformation of PdP(o-tolyl)3 groups is described as no rotor for this complex.
C,P-Palladacyclic imidate complexes for the conversion of aryl chlorides to aldehydes
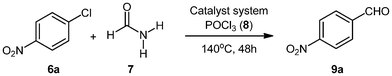
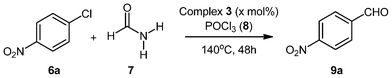
We explore here the application the new C,P-palladacyclic imidate complexes in palladium catalyzed reactions, specifically in the selective formylation of aryl halides under CO-free conditions to obtain synthetically important aromatic aldehydes.Initially, our aim was to synthesize aryl cyanides from aryl chlorides based on a recent protocol employing mild cyanide-free conditions.27 Accordingly, we submitted 4-nitrochlorobenzene to the cyanation conditions using formamide/POCl3 as the formylating reagent in the presence of different catalytic systems. Under the given conditions three types of products could form (cyanide, the amide or the aldehyde). A competing side reaction could also occur through Vilsmeier–Haack formylation of the aromatic ring at positions other than C–Cl. Our aim therefore was to selectively obtain the cyanide product over other competing side reactions in order to make the protocol synthetically useful.
At the outset of our studies the absence of palladium precursor failed to furnish any product (Table 1, entry 1), suggesting the requirement of a metal catalyst for such a transformation to take place, which was confirmed when Pd(OAc)2 was used in the absence of any activating ligand (Table 1, entry 2). Nevertheless, poor yields of the product were obtained and selectivity observed with respect to different possibilities was also low (GC-MS analysis of reaction mixture showed formation of aldehyde as the major product with small amount of cyanide and amide). Slight improvement in yield and selectivity was observed when activating phosphine ligands such as PPh3 and P(o-Tolyl)3 were employed in combination with Pd(OAc)2 as the palladium(II) precursor (Table 1, entries 3 and 4). Change in the metal to ligand ratio also had little effect on the outcome of the reaction. To ascertain the importance of POCl3 (primary reagent for the formation of Vilsmeier–Haack adduct), its exclusion from the reaction led to formation of nitrobenzene (hydro-dehalogenated product formed on hydrolysis of the oxidative addition of palladium with 4-nitrochlorobenzene). The employment of more electron-rich ligands such as SIPrHCl and X-Phos resulted in poor product yields and selectivity as compared to P(o-Tol)3 (Table 1, entries 8 and 9).
| Entry | Catalyst system (mol%) | Yieldb (%) |
|---|---|---|
| a General procedure: 6a (1 mmol), 7 (10 mL mmol−1), Pd complex, POCl3 (2.0 mmol), 140 °C, 48 h under nitrogen.b Isolated yields (product aldehydes confirmed by 1H and 13C NMR).c No POCl3 used in the reaction.d 20 mmol of POCl3 used.e Large amount of hydro-dehalogenated nitrobenzene was obtained. | ||
| 1 | — | 0 |
| 2 | Pd(OAc)2 (5) | 21 |
| 3 | Pd(OAc)2 (5)/PPh3 (10) | 28 |
| 4 | Pd(OAc)2 (5)/P(o-tol)3 (5) | 35 |
| 5 | Pd(OAc)2 (5)/P(o-tol)3 (10) | 40 |
| 6 | Pd(OAc)2 (5)/P(o-tol)3 (10) | 0c |
| 7 | Pd(OAc)2 (5)/P(o-tol)3 (10) | 42d |
| 8 | Pd(OAc)2 (5)/SIPrHCl (5) | 15 |
| 9 | Pd(OAc)2 (5)/X-Phos (5) | 12 |
| 10 | 2 (2.5) | 52 |
| 11 | 3 (2.5) | 68 |
| 12 | 4 (2.5) | 30 |
| 13 | 5 (2.5) | 27 |
| 14 | HB complex (1) (2.5) | 54 |
| 15 | POPD1 (2.5) | 36 |
| 16 | Pd(OAc)2 (5)/CM-Phos (5) | 16e |
We then employed our novel dimeric palladium complexes 2–5 and to our surprise there was a drastic improvement in the yields. On closer inspection of the products it was observed that rather than obtaining the desired cyanated product the dimeric palladium complexes led to the selective formation of the formylated 4-nitrobenzaldehyde (GCMS analysis showed exclusive formation of aldehyde product). Surprisingly, the formylation had occurred only at the C–Cl bond rather than any other position of the aromatic ring. Although the dimeric complexes have similar backbones i.e. cyclometallated phosphine coordination to the palladium centre, they differ with respect to the bridging ligand that has been employed. Next experiments were then directed to ascertain whether changes in the bridging ligand could lead to any improvement in the yields or selectivity for the above-mentioned transformation. Notable differences were observed between the imide ligands (for 2, 52%; 3, 68%; 4, 30%; 5, 27%) with the maleimide bridged palladacyclic complex 3 outperforming all others.
The commercially available Herrmann–Beller complex 1 also shows slightly reduced reactivity than 3 with the formation of the aromatic aldehyde as the major product. Commercially available CombiPhos® POPD1 dimer complex when employed also showed poor yield of the aldehyde product. Recently, CM-Phos has been shown to undergo in situ formation of a C,P-palladacycle on complexation with Pd(OAc)2.5j,28 Accordingly, CM-Phos was also subjected to the formylation conditions, however poor yields of the desired product was obtained (the major product obtained was hydro-dehalogenated nitrobenzene, Table 1, entry 16).
Based on these results we further investigated the effect of temperature on the reactivity of the palladacyclic complex 3 to catalyse the above transformation (Table 2). Initially, at a lower temperature of 100 °C complete recovery of starting material was observed (Table 2, entry 1). This suggests that the energy required for the activation of the palladacyclic complexes to efficiently catalyse the reaction cannot be achieved at this temperature. Increasing the temperature to either 150 or 170 °C resulted in drastic reduction in yields of the desired product, although this might be due to the degradation of the product formed under elevated temperature conditions (Table 2, entries 3 and 4, respectively). It is therefore evident that the optimum temperature at which the palladacyclic complex 3 efficiently catalyzes the formation of aromatic aldehydes is 140 °C.
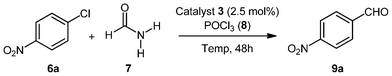
Next we turned our attention towards understanding the effect of catalyst concentration on the catalytic efficiency of palladacyclic complex 3. Catalyst loading experiments were performed to determine the overall reactivity of the complex by varying its concentration in the reaction and is generally expressed in terms of turnover number (TON = mol product/mol catalyst). Initial studies were carried out at 2.5 mol% catalyst concentration (5.0 mol% Pd) furnishing selectively the aldehyde product in 68% yield (Table 3, entry 1, TON = 27.2). A higher concentration of Pd (10.0 mol% Pd, Table 3, entry 2) led to lower yields of the formylated product (58%, TON = 11.6). Lowering of catalyst concentration to 1.25 mol% (2.5 mol% Pd, Table 3, entry 3, 39%, TON = 31.2) or 1.00 mol% (2.0 mol% Pd, Table 3, entry 4, 32%, TON = 32) resulted in drastic reduction in the product yield and selectivity. Although the turnover numbers for lower catalyst loading values (1.0 mol% and 1.25 mol%) are slightly higher than for 2.5 mol% (entry 1) the overall yield of the product is better at 2.5 mol%. Accordingly, 2.5 mol% catalyst concentration of the palladacycle 3 was selected as the optimum concentration for obtaining higher selectivity towards the formation of the aldehyde product.
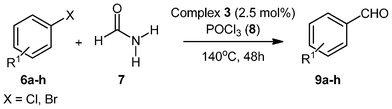
With the optimized conditions in hand, we probed the most active palladacyclic complex 3 for the formylation of further activated and unactivated aryl chlorides and bromides. Electron-withdrawing substituents such as 4-F (Table 4, entry 2), 4-CF3 (Table 4, entry 3) or 3-NO2 (Table 4, entry 4) on the aryl chloride moiety exhibited improved product yields while the unactivated chlorobenzene (Table 4, entry 5) also furnished good yield of the corresponding aldehyde. Although, enhancement in reactivity was observed for different activated (electron-attracting substituents) aryl chlorides, the same was not true for a deactivated aryl chloride such as 4-chloroanisole that gave poor yield of the desired product (Table 4, entry 6). When 1,4-dichlorobenzene was employed as substrate, a regioselectivity of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was observed for the formation of 4-chlorobenzaldehyde as compared to 1,4-benzene dicarbaldehyde (Table 4, entry 7).
:1 was observed for the formation of 4-chlorobenzaldehyde as compared to 1,4-benzene dicarbaldehyde (Table 4, entry 7).
| Entry | Aryl halide | Product | Yieldb (%) |
|---|---|---|---|
| a General procedure: aryl halide 6 (1 mmol), 7 (10 mL mmol−1), complex 3 (2.5 mol%), POCl3 (2.0 mmol), 140 °C, 48 h under nitrogen.b Isolated yields (product aldehydes confirmed by 1H and 13C NMR).c 16% of 1,4-benzene dicarbaldehyde also obtained. | |||
| 1 |  |  | 68 |
| 2 | 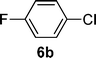 | 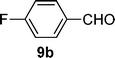 | 64 |
| 3 | 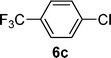 | 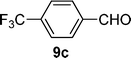 | 69 |
| 4 | 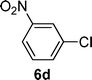 | 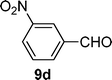 | 74 |
| 5 | 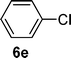 | 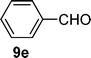 | 72 |
| 6 | 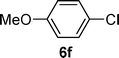 | 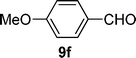 | 31 |
| 7 | 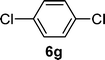 |  | 43c |
| 8 |  | 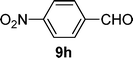 | 82 |
| 9 |  |  | — |
Aryl bromides when employed as substrates also gave high yield of the selective aromatic aldehyde exhibiting the generality of procedure towards the formylation of different aryl halides. To increase the viability of the developed protocol a more challenging ortho-substituted aryl bromide was subjected to formylation conditions. Surprisingly, the formylation reactions did not furnish any product suggesting a limitation towards sterically hindered substrates.
Suggested mechanism for palladium-catalyzed formylation of activated aryl chlorides
In case of the C,P-palladacyclic complexes the catalytically active species has been suggested to be a monoligated Pd(0) species [PdP(o-Tol)3].29 This could be generated under our specific conditions via reduction with solvent, in this case formamide.19,30 The C,P-palladacycles, as lately accepted also for other palladacycles, could be therefore considered as reservoirs of monoligated Pd(0) species and are expected to be in equilibrium with the monomeric I. Preliminary electrospray ionization studies carried out on the palladacyclic complex 3 in formamide solution also suggest the formation of the monoligated [PdP(o-Tol)3] I as the major species, although the fact that 2–5 produce such a fragment at 409 m/z on its own (see Experimental section) indicates that further experiments are needed to ascertain this observation. Additional peaks at m/z 444 and 556 detected once the reaction takes place makes the following mechanism plausible. The first step of the suggested catalytic cycle (Fig. 2) would involve the oxidative addition of the aryl chloride (6e) with the monomeric palladium(0) species I resulting in the formation of a 14-electron monoligated species II. Transmetallation with the in situ generated Vilsmeier reagent A would give intermediate III. Reductive elimination will then generate the imine product which on hydrolysis would give the aldehyde 9e. The catalytically active monoligated species I would then be regenerated by the loss of HCl from species IV, thus re-entering the catalytic cycle.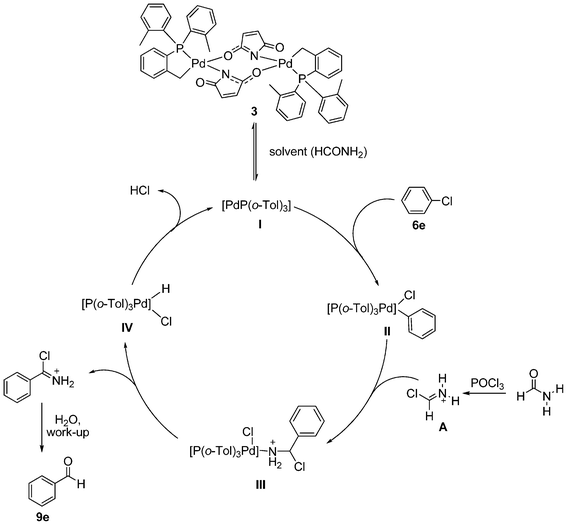 | ||
| Fig. 2 Proposed mechanistic pathway for the palladium-catalyzed formylation of activated aryl chlorides. | ||
Conclusions
By means of simple acid–base reaction with imidate ligands new phosphapalladacyclic complexes with interesting catalytic properties have been prepared. Extensive characterization has been carried out, including X-ray single-crystal analysis. Their application in the efficient and selective formylation of activated aryl chlorides has been explored and presented in this report.Experimental section
General remarks
C, H and N analyses were carried out with a Carlo Erba instrument. IR spectra were recorded on a Perkin-Elmer spectrophotometer 16F PC FT-IR, using Nujol mulls between polyethylene sheets. NMR data (1H) were recorded on Bruker 200, 300 or 400 spectrometers. Mass spectrometric analyses were performed on a Fisons VG Autospec double-focusing spectrometer, operated in positive mode. Ions were produced by fast atom bombardment (FAB) with a beam of 25-keV Cs atoms. The mass spectrometer was operated with an accelerating voltage of 8 kV and a resolution of at least 1000. The cyclometallated precursor [{Pd(μ-AcO)(CˆP)}2] 1 was prepared as described previously.4c The commercially available chemicals were purchased from Aldrich Chemical Co. and were used without further purification, and all the solvents were dried by standard methods before use.Synthesis
:2). A clear solution was immediately obtained, and it was stirred at reflux temperature for 1 h. Then it was concentrated under reduced pressure until ca. one fifth of the initial volume. Slow addition of diethyl ether caused the formation of the complexes, which were filtered off, washed with diethyl ether and air-dried. The compounds were recrystallised from dichloromethane-hexane.[{Pd(μ-suc)(CˆP)}2] 2. (0.243 g, 75%). Anal. Calc. for C50H48N2O4P2Pd2: C 59.1; H 4.7; N 2.7. Found: C 59.3; H 4.9; N 2.8%. IR (cm−1): 1724s, 1600 vs, 758 s, 584 s, 526 s, 466 s, 456 s. 1H NMR (300 MHz; CD3CN, 298 K, δ): 7.20–6.70 (m, br, 24 H, arom), 3.32 (s, br, 4 H, CH2 benzyl), 2.67 (s, br, 12 H, CH3 benzyl), 2.45 (s, br, 8 H, CH2 suc). 31P NMR (300 MHz; CD3CN, 298 K, δ): 34.9. FAB-MS (positive mode) m/z: 918 [{Pd(CˆP)}2(suc)], 713 [Pd(CˆP)2]+, 508 [{Pd(suc)(CˆP)}]+, 409 [Pd(CˆP)]+.
[{Pd(μ-mal)(CˆP)}2] 3. (0.223 g, 69%). Anal. Calc. for C50H44N2O4P2Pd2: C 59.3; H 4.4; N 2.7. Found: C 59.4; H 4.5; N 2.9%. IR (cm−1): 1724 s, 1614 vs, 1586 s, 756 s, 584 s, 526 s, 468 s, 460 s. 1H NMR (300 MHz; CD3CN, 298 K, δ): 7.00–6.64 (m, br, 24 H, arom), 6.49 (s, 4 H, mal), 3.41 (s, br, 4 H, CH2 benzyl), 2.68 (s, br, 12 H, CH3 benzyl). 31P NMR (300 MHz; CD3CN, 298 K, δ): 35.5. FAB-MS (positive mode) m/z: 916 [{Pd(CˆP)}2(mal)], 504 [{Pd(mal)(CˆP)}]+, 409 [Pd(CˆP)]+.
[{Pd(μ-phthal)(CˆP)}2] 4. (0.249 g, 70%). Anal. Calc. for C58H48N2O4P2Pd2: C 62.7; H 4.3; N 2.5. Found: C 62.8; H 4.4; N 2.7%. IR (cm−1): 1728 s, 1632 vs, 1620 s, 758 s, 586 s, 526 s, 480 s, 448 s. 1H NMR (300 MHz; CD3CN, 298 K, δ): 7.31–6.76 (m, br, 32 H, arom), 3.41 (s, br, 4 H, CH2 benzyl), 2.73 (s, br, 12 H, CH3 benzyl). 31P NMR (300 MHz; CD3CN, 298 K, δ): 35.5. FAB-MS (positive mode) m/z: 966 [{Pd(CˆP)}2(phthal)], 556 [{Pd(phthal)(CˆP)}]+, 409 [Pd(CˆP)]+.
[{Pd(μ-sacc)(CˆP)}2] 5. (0.306 g, 81%). Anal. Calc. for C56H48N2O6P2S2Pd2: C 56.8; H 4.1; N 2.4. Found: C 57.0; H 4.3; N 2.5%. IR (cm−1): 1719 s, 1627 vs, 1583 s, 755 s, 594 s, 528 s, 470 s, 453 s.1H NMR (300 MHz; CD3CN, 298 K, δ): 7.30–6.61 (m, br, 32 H, arom), 3.50 (s, br, 4 H, CH2 benzyl), 2.73 (s, br, 12 H, CH3 benzyl). 31P NMR (300 MHz; CD3CN, 298 K, δ): 35.3. FAB-MS (positive mode) m/z: 1000 [{Pd(CˆP)}2(sacc)], 591 [{Pd(sacc)(CˆP)}]+, 409 [Pd(CˆP)]+.
4-Nitrobenzaldehyde. 1H NMR (300 MHz, DMSO-d6): δ 10.20 (s, 1 H), 8.48 (d, J = 7.6 Hz, 2 H), 8.18 (d, J = 7.6 Hz, 2 H); 13C NMR (75 MHz, DMSO-d6): δ 192.3, 140.0, 130.6, 124.2, 122.3; MS (EI) m/z (relative intensity) 151 (100) [M+]. The spectral data were in accordance with those reported in the literature.31
4-Fluorobenzaldehyde. 1H NMR (300 MHz, DMSO-d6): δ 10.00 (s, 1 H), 7.94 (td, J = 5.7, 2.1 Hz, 2 H), 7.19 (t, J = 8.3 Hz, 2 H); 13C NMR (75 MHz, DMSO-d6): δ 191.2, 167.4, 133.4, 132.2, 116.6; MS (EI) m/z (relative intensity) 125 (100) [M+]. The spectral data were in accordance with those reported in the literature.31
4-Trifluoromethylbenzaldehyde. 1H NMR (300 MHz, DMSO-d6): δ 10.13 (s, 1 H), 8.08 (d, J = 8.1 Hz, 2 H), 7.72 (d, J = 7.9 Hz, 2 H); 13C NMR (75 MHz, DMSO-d6): δ 191.7, 138.7, 133.8, 129.8, 126.0, 125.8; MS (EI) m/z (relative intensity) 174 (100) [M+]. The spectral data were in accordance with those reported in the literature.31
3-Nitrobenzaldehyde. 1H NMR (300 MHz, DMSO-d6): δ 10.26 (s, 1 H), 8.14–8.09 (m, 1 H), 7.88–7.83 (m, 3 H); 13C NMR (75 MHz, DMSO-d6): δ 190.3, 149.6, 134.7, 134.6, 131.0, 130.3, 124.8; MS (EI) m/z (relative intensity) 151 (100) [M+]. The spectral data were in accordance with those reported in the literature.31
Benzaldehyde. 1H NMR (300 MHz, DMSO-d6): δ 10.03 (s, 1 H), 7.98 (d, J = 7.0 Hz, 2 H), 7.65 (d, J = 7.3 Hz, 1 H), 7.56 (t, J = 7.5 Hz, 2 H); 13C NMR (75 MHz, DMSO-d6): δ 193.2, 136.5, 136.1, 129.7, 129.2; MS (EI) m/z (relative intensity) 106 (100) [M+]. The spectral data were in accordance with those reported in the literature.31
4-Methoxybenzaldehyde. 1H NMR (300 MHz, CDCl3): δ 10.06 (s, 1 H), 8.01 (d, J = 8.2 Hz, 2 H), 7.12 (d, J = 8.2 Hz, 2 H), 4.03 (s, 3 H); 13C NMR (75 MHz, CDCl3): δ 190.8, 164.6, 132.0, 130.0, 114.3, 55.6; MS (EI) m/z (relative intensity) 136 (100) [M+]. The spectral data were in accordance with those reported in the literature.31
4-Chlorobenzaldehyde. 1H NMR (300 MHz, DMSO-d6): δ 10.14 (s, 1 H), 8.11 (d, J = 8.0 Hz, 2 H), 7.69 (d, J = 8.0 Hz, 2 H); 13C NMR (75 MHz, DMSO-d6): δ 192.6, 139.8, 135.3, 130.8, 129.2; MS (EI) m/z (relative intensity) 140 (100) [M+]. The spectral data were in accordance with those reported in the literature.32
| Empirical formula | C53H54N2O5P2Pd2 |
| M | 1073.72 |
| T/K | 100(2) |
| λ/Å | 0.71073 |
| Crystal system | Triclinic |
| Space group | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a/Å | 13.2632(10) |
| b/Å | 13.7584(10) |
| c/Å | 15.9030(12) |
| α/° | 102.9940(10) |
| β/° | 92.4610(10) |
| γ/° | 113.4320(10) |
| V/Å3 | 2565.9(3) |
| Z | 2 |
| Dc/Mg m−3 | 1.390 |
| μ/mm−1 | 0.809 |
| F(000) | 1096 |
| θ range for data collection/° | 1.33–28.63 |
| Reflections collected | 31383 |
| Independent reflections | 12002 |
| Data/parameters/restrains | 12002/0/562 |
| Goodness-of-fit on F2 | 1.082 |
| Final R indices [I > 2σ(I)] | R1 = 0.0518, wR2 = 0.1702 |
| R indices (all data) | R1 = 0.0562, wR2 = 0.1764 |
| Max./min. Δρ/e Å−3 | 3.165, −1.019 |
Acknowledgements
We thank Fundación Séneca (project 08670/PI/08) for financial support and Department of Science and Technology-India for a Fast Track fellowship to A. R. K. (SR/FT/CS-58/2010). This work was partially supported by CICYT-MICINN (CTQ 2011-28903-C02).References
- (a) J. P. Kleinman and M. Dubeck, J. Am. Chem. Soc., 1963, 85, 1544 CrossRef; (b) A. C. Cope and R. W. Siekman, J. Am. Chem. Soc., 1965, 87, 3273 Search PubMed; (c) A. Kasahara, Bull. Chem. Soc. Jpn., 1968, 41, 1272 CrossRef CAS.
- (a) I. Omae, Coord. Chem. Rev., 1988, 83, 137 CrossRef CAS; (b) M. Ghedini, I. Aiello, A. Crispini, A. Golemme, M. La Deda and D. Pucci, Coord. Chem. Rev., 2006, 250, 1373 CrossRef CAS; (c) I. Omae, J. Organomet. Chem., 2007, 692, 2608 CrossRef CAS; (d) K. Gogula and D. Sames, Science, 2006, 312, 67 CrossRef; (e) J. Dupont, C. S. Consorti and J. Spencer, Chem. Rev., 2005, 105, 2527 CrossRef CAS; (f) Palladacycles, ed. J. Dupont and M. Pfeffer, Wiley-VCHWeinheim, 2008 Search PubMed; (g) D. A. Alonso and C. Nájera, Chem. Soc. Rev., 2010, 39, 2891 RSC; (h) G. R. Peh, E. A. B. Kantchev, J. C. Er and J. Y. Ying, Chem.–Eur. J., 2010, 16, 4010 CrossRef CAS.
- (a) A. Herrman, V. P. W. Böhm and C.-P. Reisinger, J. Organomet. Chem., 1999, 576, 23 CrossRef; (b) R. B. Bedford, Chem. Commun., 2003, 1787 RSC.
- (a) W. A. Herrmann, C. Brossmer, K. Öfele, C.-P. Reisinger, T. Piermeier, M. Beller and H. Fischer, Angew. Chem., 1995, 107, 1989 CrossRef; (b) W. A. Herrmann, C. Brossmer, K. Öfele, C.-P. Reisinger, T. Piermeier, M. Beller and H. Fischer, Angew. Chem., Int. Ed. Engl., 1995, 34, 1844 CrossRef CAS; (c) W. A. Herrmann, C. Brossmer, C.-P. Reisinger, T. H. Riermeier, K. Öfele and M. Beller, Chem.–Eur. J., 1997, 3, 1357 CrossRef CAS.
- (a) W. A. Herrmann, K. Öfele, D. v. Preysing and S. K. Schneider, J. Organomet. Chem., 2003, 687, 229 CrossRef CAS; (b) V. Farina, Adv. Synth. Catal., 2004, 346, 1553 CrossRef CAS; (c) K. Yu, W. Sommer, J. M. Richardson, M. Weck and C. W. Jones, Adv. Synth. Catal., 2005, 347, 161 CrossRef CAS; (d) Ch. Xu, G. J. H. Kennard, F. Hennersdorf, Y. Li, S. A. Pullarkat and P.-H. Leung, Organometallics, 2012, 31, 3022 CrossRef CAS; (e) T. Mizuta, N. Tanaka, Y. Iwakuni, K. Kubo and K. Miyoshi, Organometallics, 2009, 28, 2808 CrossRef CAS; (f) M. E. Günay and Ch. J. Richards, Organometallics, 2009, 28, 5833 CrossRef; (g) O. N. Gorunova, M. V. Livantsov, Y. K. Grishin, N. A. Kataeva, K. A. Kochetkov, A. V. Churakov, L. G. Kuz'mina and V. V. Dunina, Polyhedron, 2012, 31, 37 CrossRef CAS; (h) D. E. Janzen, D. G. VanDerveer, L. F. Mehne, D. A. da Silva Filho, J.-L. Brédas and G. J. Grant, Dalton Trans., 2008, 1872 RSC; (i) V. A. Stepanova, L. M. Egan, L. Stahl and I. P. Smoliakova, J. Organomet. Chem., 2011, 696, 3162 CrossRef CAS; (j) C. M. So, C. P. Lau and F. Y. Kwong, Angew. Chem., Int. Ed., 2008, 47, 8059 CrossRef CAS.
- (a) M. Beller, T. H. Riermeier, W. Mägerlein, T. E. Müller and W. Scherer, Polyhedron, 1998, 17, 1165 CrossRef CAS; (b) B. L. Shaw, S. D. Perera and E. A. Staley, Chem. Commun., 1998, 1361 RSC; (c) G. D. Frey, C.-P. Reisinger, e. Herdtweck and W. A. Herrmann, J. Organomet. Chem., 2005, 690, 3193 CrossRef CAS.
- (a) J. L. Serrano, L. García, J. Pérez, E. Pérez, G. Sánchez, J. García, P. Sehnal, S. de Ornellas, T. Williams and I. J. S. Fairlamb, Organometallics, 2011, 30, 5095 CrossRef CAS; (b) M. D. Santana, R. García-Bueno, G. García, G. Sánchez, J. García, J. Pérez, L. García and J. L. Serrano, Dalton Trans., 2011, 40, 3537 RSC; (c) M. D. Santana, R. García-Bueno, G. García, G. Sánchez, J. García, J. Pérez, L. García and J. L. Serrano, Inorg. Chim. Acta, 2011, 378, 49 CrossRef CAS; (d) J. L. Serrano, L. García, J. Pérez, E. Pérez, J. M. Galiana, J. García, M. Martínez, G. Sánchez and I. da Silva, Dalton Trans., 2011, 40, 156 RSC; (e) J. Pérez, A. Espinosa, J. M. Galiana, E. Pérez, J. L. Serrano, M. A. G. Aranda and M. Insausti, Dalton Trans., 2009, 9625 RSC; (f) J. L. Serrano, L. García, J. Pérez, E. Pérez, J. García, G. Sánchez, G. López and M. Liu, Eur. J. Inorg. Chem., 2008, 4797 CrossRef CAS; (g) J. Pérez, J. L. Serrano, J. M. Galiana, F. L. Cumbrera, A. L. Ortiz, G. Sánchez and J. García, Acta Crystallogr., Sect. B: Struct. Sci., 2007, 63, 75 Search PubMed; (h) G. Sánchez, J. Vives, G. López, J. L. Serrano, L. García and J. Pérez, Eur. J. Inorg. Chem., 2005, 2360 CrossRef; (i) G. Sánchez, J. García, D. Meseguer, J. L. Serrano, L. García, J. Pérez and G. López, Dalton Trans., 2003, 4709 RSC.
- (a) J. L. Serrano, I. J. S Fairlamb, G. Sánchez, L. García, J. Pérez, J. Vives, G. López, C. M. Crawforth and R. J. K. Taylor, Eur. J. Inorg. Chem., 2004, 2706 CrossRef CAS; (b) G. Sánchez, J. L. Serrano, J. García, G. López, J. Pérez and E. Molins, Inorg. Chim. Acta, 1999, 287, 37 CrossRef; (c) G. Sánchez, J. L. Serrano, J. Pérez, M. C. Ramírez de Arellano, G. López and E. Molins, Inorg. Chim. Acta, 1999, 295, 136 CrossRef.
- I. J. S Fairlamb, A. R. Kapdi, A. F. Lee, G. Sánchez, G. López, J. L. Serrano, L. García, J. Pérez and E. Pérez, Dalton Trans., 2004, 3970 RSC.
- C. M. Crawforth, I. J. S Fairlamb, A. R. Kapdi, J. L. Serrano, R. J. K. Taylor and G. Sánchez, Adv. Synth. Catal., 2006, 348, 405 CrossRef CAS.
- (a) J. Matsunami, H. Urata and K. Matsumoto, Inorg. Chem., 1995, 34, 202 CrossRef CAS; (b) J. Berners Price Susan, M. J. DiMartino, D. T. Hill, R. Kuroda, M. A. Mazid and P. J. Sadler, Inorg. Chem., 1985, 24, 3425 CrossRef; (c) J. Ruiz, J. Lorenzo, L. Sanglas, N. Cutillas, C. Vicente, M. D. Villa, F. X. Aviles, G. Lopez, V. Moreno, J. Perez and D. Bautista, Inorg. Chem., 2006, 45, 6347 CrossRef CAS.
- (a) H. M. Colquhoum, R. A. Fairman, P. Tootell and D. J. Williams, J. Chem. Soc., Dalton Trans., 1999, 2651 RSC; (b) N. Ichieda, T. Kamimura, S. Yamaguchi, Y. Funahashi, T. Ozawa, K. Jitsukawa and H. Masuda, Adv. Mater. Res., 2006, 11–12, 473 CrossRef CAS.
- (a) E. J. Baran and V. T. Yilmaz, Coord. Chem. Rev., 2006, 250, 1980 CrossRef CAS; (b) E. Guney, V. T. Yilmaz and O. Buyukgungor, Inorg. Chim. Acta, 2010, 363, 2416 CrossRef CAS; (c) E. Guney, V. T. Yilmaz and C. Kazak, Polyhedron, 2010, 29, 1285 CrossRef CAS; (d) E. Guney, V. T. Yilmaz and O. Buyukgungor, Polyhedron, 2011, 30, 1968 CrossRef CAS.
- (a) F. Aldabbagh, Compr. Org. Funct. Group Transform. II, 2005, 3, 99 CrossRef CAS; (b) L. P. Crawford and S. K. Richardson, General and Synthetic Methods, 1994, 16, 37 CAS.
- (a) W. MNgerlein, A. F. Indolese and M. Beller, J. Organomet. Chem., 2002, 641, 30 CrossRef; (b) M. Beller and A. F. Indolese, Chimia, 2001, 55, 684 CAS; (c) A. Schnyder, M. Beller, G. Mehltretter, A. F. Indolese, T. Nsenda and M. Studer, J. Org. Chem., 2001, 66, 4311 CrossRef CAS; (d) W. MNgerlein, A. Indolese and M. Beller, Angew. Chem., Int. Ed., 2001, 40, 2856 CrossRef; (e) W. Mngerlein, A. Indolese and M. Beller, J. Mol. Catal. A: Chem., 2000, 156, 213 CrossRef; (f) S. Klaus, H. Neumann, A. Zapf, D. Strabing, S. Habner, J. Almena, T. Riermeier, P. Groß, M. Sarich, W. Krahnert, K. Rossen and M. Beller, Angew. Chem., Int. Ed., 2006, 45, 154 CrossRef CAS; (g) Y. Ben-David, M. Portno and D. Milstein, J. Chem. Soc., Chem. Commun., 1989, 1816 RSC.
- V. P. Baillargeon and J. K. Stille, J. Am. Chem. Soc., 1986, 108, 452 CrossRef CAS.
- R. Mutin, C. Lucas, J. Thivole-Cazat, V. Dufaud, F. Dany and J. M. Basset, J. Chem. Soc., Chem. Commun., 1988, 896 RSC.
- (a) A. Suzuki, J. Organomet. Chem., 1999, 576, 147 CrossRef CAS; (b) A. Zapf, in Transition Metals for Organic Synthesis: Building Blocks and Fine Chemicals, Wiley-VCH, Weinheim, 2004 Search PubMed; (c) K. Tamao, K. Sumitani and M. Kumada, J. Am. Chem. Soc., 1972, 94, 4374 CrossRef CAS; (d) J. K. Stille, Angew. Chem., Int. Ed. Engl., 1986, 25, 508 CrossRef; (e) J. F. Hartwig, Acc. Chem. Res., 1998, 31, 852 CrossRef CAS; (f) B. H. Yang and S. L. Buchwald, J. Organomet. Chem., 1999, 576, 125 CrossRef CAS.
- R. B. Bedford, C. S. J. Cazin and D. Holder, Coord. Chem. Rev., 2004, 248, 2283 CrossRef CAS.
- (a) A. J. Cheney and B. L. Shaw, J. Chem. Soc., Dalton Trans., 1972, 754 RSC; (b) E. C. Alyea, S. A. Dias, G. Ferguson and P. J. Roberts, J. Chem. Soc., Dalton Trans., 1979, 948 RSC; (c) T. Mitsudo, W. Fischetti and R. F. Heck, J. Org. Chem., 1984, 49, 1640 CrossRef CAS.
- J. Forniés, A. Martín, R. Navarro, V. Sicilia and P. Villarroya, Organometallics, 1996, 15, 1826 CrossRef.
- H. Adams, N. A. Bailey, T. N. Briggs, J. A. McCleverty, H. M. Colquhoun and D. J. Williams, J. Chem. Soc., Dalton Trans., 1986, 813 RSC.
- M. Kessler, J. Pérez, M. C. Bueso, L. García, E. Pérez, J. L. Serrano and R. Carrascosa, Acta Crystallogr., Sect. B: Struct. Sci., 2007, 63, 869 CAS.
- J. Pérez, L. García, E. Pérez, J. L. Serrano, J. F. Martínez, G. Sánchez, G. López, A. Espinosa, M. Liu and F. Sanz, New J. Chem., 2003, 27, 1490 RSC.
- (a) A. L. Rheingold and C. Fultz W, Organometallics, 1984, 3, 1414 CrossRef CAS; (b) A. J. Canty, J. L. Hoare, B. W. Skelton, A. H. White and G. van Koten, J. Organomet. Chem., 1998, 552, 23 CrossRef CAS; (c) G. D. Frey, J. Schultz, E. Herdtweck and W. A. Herrmann, Organometallics, 2005, 24, 4416 CrossRef CAS; (d) J. Louie and J. F. Hartwig, Angew. Chem., Int. Ed. Engl., 1996, 35, 2359 CrossRef CAS.
- I. Dance and M. Scudder, J. Chem. Soc., Dalton Trans., 2000, 1579 RSC.
- D. N. Sawant, Y. S. Wagh, P. J. Tambade, K. D. Bhatte and B. M. Bhanage, Adv. Synth. Catal., 2011, 353, 781 CrossRef CAS.
- C. M. So and F. Y. Kwong, Chem. Soc. Rev., 2011, 40, 4963 RSC.
- (a) M. S. Drive and and J. F. Hartwig, J. Am. Chem. Soc., 1997, 119, 8232 CrossRef; (b) J. F. Hartwig, S. Richards, D. Barañano and F. Paul, J. Am. Chem. Soc., 1996, 118, 3626 CrossRef CAS; (c) R. A. Widenhoefer and S. L. Buchwald, Organometallics, 1996, 15, 2755 CrossRef CAS.
- F. d Orlye and A. Jutand, Tetrahedron, 2005, 61, 9670 CrossRef CAS.
- L. J. Gooßen, B. A. Khan, T. Fett and M. Treu, Adv. Synth. Catal., 2010, 352, 2166 CrossRef.
- A. Wang and H. Jiang, J. Org. Chem., 2010, 75, 2321 CrossRef CAS.
- SAINT, Version 6.22, Bruker AXS Inc Search PubMed.
- G. M. Sheldrick, SADABS, University of Göttingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, SHELX-97. Programs for Crystal Structure Analysis (Release 97-2), University of Göttingen, Germany, 1998 Search PubMed.
Footnote |
| † CCDC reference number 895458. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra21770h |
| This journal is © The Royal Society of Chemistry 2012 |