White light from dispersible lanthanide-doped LaVO4 core–shell nanoparticles†
Saurabh
Singh
a,
Anurodh
Tripathi
a,
Chandresh
Kumar Rastogi
b and
Sri
Sivakumar
*abc
aDepartment of Chemical Engineering, Indian Institute of Technology Kanpur, Kanpur-208016, India. E-mail: srisiva@iitk.ac.in
bMaterials Science Programme, Indian Institute of Technology Kanpur, Kanpur-208016, India
cCentre for Environmental Science and Engineering, Indian Institute of Technology Kanpur, Kanpur-208016, India
First published on 4th October 2012
Abstract
White light has been generated from dispersible lanthanide ion-doped LaVO4 core–shell nanoparticles by using a single UV excitation source. The core–shell nanoparticles are excited with single wavelength 280 nm UV light, in which the energy transfer from [VO4]3− groups to Ln3+ ions occurs to produce bright white light. The absolute quantum yields of LaVO4:Tm3+/Tb3+/Eu3+ (Type 1) and LaVO4:Tm3+/Dy3+ (Type 2) core–shell nanoparticles have been estimated as 5% and 7% respectively. The control core nanoparticles doped with all the lanthanide ions did not produce white light which proves the need of a core–shell structure. The core–shell structure of LaVO4 nanoparticles has been investigated by TEM, EDX, XRF, and PL spectroscopy techniques.
Introduction
Nanocrystalline phosphors attract a great deal of interest for the generation of cheap and efficient white light for applications such as display devices, general lighting, and active materials in lasers.1–3 The devices that have been developed to produce white light include incandescent lamps, fluorescent light tubes, compact fluorescent lamps, etc. and the latest additions to the field are organic LED's and inorganic LED's which exploit electricity for light generation.4–11 In these devices, white light has been generated either by mixing three different monochromatic sources (RGB) or conversion of UV/blue light by using phosphors.12 These devices suffer either from single or multiple issues such as long term stability of emitters, high cost, and low efficiency. For the three converter (RGB) system, the manufacture cost is high and the blue emission intensity is poor because of strong reasbsorption of blue light by the red and green-emitting ions.13,14 Therefore, to circumvent these disadvantages there is a need to produce a single phase material that would be more stable, efficient, and durable as well as cost effective. To this end, lanthanide ion-doped nanomaterials are gaining popularity due to their increased photostability, sharp emission lines, large stokes shift, and longer lifetime.15–17 They can produce light by three photophysical processes: 1) up-conversion18–20 (conversion of two or more lower energy photons into one higher energy photon), 2) conventional ‘stokes’ shift fluorescence,21 (conversion of one higher energy photon into one lower energy photon), and, 3) quantum cutting22,23 (conversion of one higher energy photon into two or more lower energy photons). There are reports available on the generation of white light from 1) up-conversion nanoparticles,24 2) multielement (Ln3+)-doped core nanomaterials,25 3) lanthanide-doped bulk materials,26,27 and 4) lanthanide-complexes.28 Few reports are available on the generation of white light from lanthanide-doped nanomaterials through up-conversion,29–31 however, the efficiency is very low (<0.5%).29,32 Though multielement-doped nanomaterials produce white light, their efficiency can be limited by the internal energy transfer between them. Additionally, processing will be a challenging task for lanthanide-doped bulk materials in fabrication of devices. It is well known that lanthanide complexes have inferior optical properties compared to lanthanide-doped inorganic matrices. Furthermore, there are a number of reports available on the generation of white, red, green and blue light by conventional ‘stokes’ shift fluorescence because this process is more efficient than up-conversion.22,33 However, they have several limitations such as, inefficient direct excitation of lanthanide ions, internal energy transfer due to multiple dopant in a core matrix, poor dispersibility in organic solvents, use of multiple phosphors, multiple excitation sources, etc.34 To our knowledge, nobody has so far reported the generation of white light through ‘stokes’ shift fluorescence from a single type dispersible lanthanide-doped core nanoparticle by using a single excitation source (e.g. UV light).In this article, we report the generation of white light from dispersible lanthanide ion-doped LaVO4 core–shell nanoparticles via a ‘stokes’ shift fluorescence process by using a single excitation source (UV light). We note that direct excitation of lanthanide ions is an inefficient process due to the lesser extinction coefficient (1–10 M−1 cm−1) compared to energy transfer from a host material (LnVO4, LnPO4, TiO2, etc.) or other ions13,15 (Ce3+ and Yb3+).35–40 The vanadate charge transfer transition has several orders of magnitude of extinction coefficient and a broad absorption range (260–340 nm) compared to lanthanide ions.36 Additionally, thermally activated energy migration from [VO4]3− to lanthanide ions is very efficient and it has been universally used as a sensitizer for most of the lanthanide ions.41
Experimental
Materials
All the lanthanide salts and sodium orthovanadate were obtained from Sigma–Aldrich. Ethanol was obtained from Merck's chemicals and dichloromethane, oleic acid, and triethylamine were obtained from Qualigens India Ltd. All the chemicals and salts were used without any further purification.Preparation of oleic acid stabilized lanthanide-doped LaVO4 core–shell nanoparticles
Oleic acid (3 ml) was dissolved in 70 ml of distilled water–ethanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture. The pH of the solution was adjusted to 6 by adding a few drops of triethylamine (30% w/v) and the solution was heated to 75 °C. The total stoichiometric amount (6 mmol for type 1 and 4 mmol for type 2 core–shell particles) of aqueous solution of sodium orthovanadate (3 mL) was added dropwise to the above solution, followed by the dropwise addition of 2 ml aqueous solution of La(NO3)3 and Tm(NO3) salts (total 1.33 mmol). The mixture was stirred for 10 min to prepare the core nanoparticles followed by the dropwise addition of 2 ml aqueous solution of La(NO3)3 salts (total 1.33 mmol) to prepare the inert shell. The mixture was again stirred for 10 min and subsequent shells were prepared by adding Ln(NO3)3 salt after every 10 min to prepare the shells. After the addition of all the lanthanide salt portions, the mixture was allowed to stir for 2 h and the precipitate was washed with water and ethanol to remove the excess oleic acid. The type 1 core–shell particles consist of Tm3+ ions (20%) in the LaVO4 core, Tb3+ (10%) and Eu3+ (0.5%) ions in the second and fourth LaVO4 shell respectively. The first, third and fifth shells are undoped LaVO4 shell. In type 2 core-shell particles, Tm3+ (15%) and Dy3+ (0.5%) ions are doped in the LaVO4 core and second LaVO4 shell, respectively, whereas the first and third shells are undoped LaVO4 shell.
:1) mixture. The pH of the solution was adjusted to 6 by adding a few drops of triethylamine (30% w/v) and the solution was heated to 75 °C. The total stoichiometric amount (6 mmol for type 1 and 4 mmol for type 2 core–shell particles) of aqueous solution of sodium orthovanadate (3 mL) was added dropwise to the above solution, followed by the dropwise addition of 2 ml aqueous solution of La(NO3)3 and Tm(NO3) salts (total 1.33 mmol). The mixture was stirred for 10 min to prepare the core nanoparticles followed by the dropwise addition of 2 ml aqueous solution of La(NO3)3 salts (total 1.33 mmol) to prepare the inert shell. The mixture was again stirred for 10 min and subsequent shells were prepared by adding Ln(NO3)3 salt after every 10 min to prepare the shells. After the addition of all the lanthanide salt portions, the mixture was allowed to stir for 2 h and the precipitate was washed with water and ethanol to remove the excess oleic acid. The type 1 core–shell particles consist of Tm3+ ions (20%) in the LaVO4 core, Tb3+ (10%) and Eu3+ (0.5%) ions in the second and fourth LaVO4 shell respectively. The first, third and fifth shells are undoped LaVO4 shell. In type 2 core-shell particles, Tm3+ (15%) and Dy3+ (0.5%) ions are doped in the LaVO4 core and second LaVO4 shell, respectively, whereas the first and third shells are undoped LaVO4 shell.
Characterizations
The photoluminescence spectra and decay curves were recorded using an Edinburgh instruments FLSP 920 fluorescence system equipped with a red-sensitive Peltier element cooled Hamamatsu R928-P PMT. Emission and excitation spectra were measured by exciting the samples with a 450 W steady state Xe lamp. Decay curves were measured with a Nd:YAG laser, attached to an optical parametric oscillator (OPO) with an optical range from 210–2400 nm. Absolute quantum yields were measured with an integrating sphere coated with BaSO4. The TEM images were obtained from FEI Technai G2 U-Twin (200 KeV) instrument. 1H NMR and FTIR spectra were measured by using a Bruker AC 300 and Bruker Alfa respectively. XRD analyses were done using a Siemens D5000 Bragg-Brentano θ–2θ diffractometer. The nanoparticles (20–25 mg) were smeared onto a zero-diffraction quartz plate using ethanol. Step-scan X-ray powder-diffraction data were collected over the 2θ range 3–100° with Cu-Kα (40 kV, 40 mA) radiation on a Siemens D5000 Bragg-Brentano θ–2θ diffractometer equipped with a diffracted-beam graphite monochromator crystal, 2 mm (1°) divergence and anti-scatter slits, 0.6 mm receiving slit, and incident beam Soller slit. The scanning step size was 0.04° 2θ with a counting time of 1.5 s step−1. X-ray fluorescence spectroscopy data were obtained from a ZSX primus series, Rigaku corporation spectrometer. EDAX analysis was done with a Carl Zeiss NTS GmbH-SUPRA, 40 VP FESEM.Results and discussion
The white light has been generated from two types of core–shell nanoparticles: The type 1 core–shell nanoparticle is doped with Tm3+ (blue)/Tb3+ (green)/Eu3+ (red) ions and type 2 is doped with Tm3+ (blue)/Dy3+ (yellow) ions in different LaVO4 shells, separated by an inert (un-doped) LaVO4 layer (Fig. 1a). The core–shell nanoparticles are excited with single wavelength 280 nm UV light, in which the energy transfer from [VO4]3− groups to Ln3+ ions is exploited to produce bright white light.41 Moreover, these core–shell nanoparticles can be dispersible in organic solvents and polymers, which can easily be processed via spin-coating for flat panel displays and LEDs applications. The CIE co-ordinates of the light emitted from the nanoparticles can also be changed by adjusting the concentration and ratio of lanthanide ions doped in different shells. Furthermore, these particles can be excited with a broad range of UV wavelength (260–340 nm) which is advantageous to general lighting applications.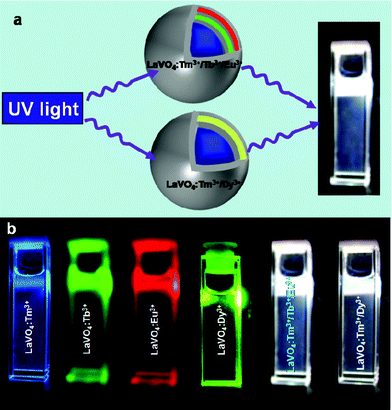 | ||
| Fig. 1 a) Schematic representation of the generation of white light from LaVO4:Tm3+/Tb3+/Eu3+ (Type 1) and LaVO4:Tm3+/Dy3+ (Type 2) core–shell nanoparticles and b) digital photographs of blue, green, red, yellow and white light emissions from colloidal dispersions of LaVO4:Tm3+, LaVO4:Tb3+, LaVO4:Eu3+, LaVO4:Dy3+ core nanoparticles and LaVO4:Tm3+/Tb3+/Eu3+ (Type 1) and LaVO4:Tm3+/Dy3+ (Type 2) core–shell nanoparticles in dichloromethane solvent (1 mg mL−1). The samples were excited with a 280 nm Xe lamp light. | ||
The LaVO4 core–shell particles stabilized with oleic acid were prepared by the co-precipitation method. Fig. 1a shows the schematic representation of type 1 (LaVO4:Tm3+/Tb3+/Eu3+) and type 2 (LaVO4:Tm3+/Dy3+) LaVO4 core–shell nanoparticles. In type 1, Tm3+ ions (20%) are doped in the LaVO4 core, Tb3+ (10%) and Eu3+ (0.5%) ions are doped in the second and fourth LaVO4 shell respectively. The first, third and fifth shells are inert shell (un-doped), which separate the lanthanide-ions from each other to prevent quenching due to internal energy transfer. The outer inert shell also prevents quenching from environmental effects such as solvents and OH groups. In type 2 Tm3+ (15%) and Dy3+ (0.5%) ions are doped in the LaVO4 core and second LaVO4 shell, respectively, whereas the first and third shells are inert shells. Fig. 1b shows the digital photographs of bright white light emissions from LaVO4:Tm3+/Tb3+/Eu3+ (Type 1), and LaVO4:Tm3+/Dy3+ (Type 2) core–shell nanoparticles dispersed in dichloromethane by excitation with 280 nm light. Additionally, bright blue, green, red, and yellow emissions have been observed by excitation with 280 nm light from LaVO4:Tm3+, LaVO4:Tb3+, LaVO4:Eu3+, LaVO4:Dy3+ core nanoparticles, respectively. The digital photographs clearly suggest that the bright white light from both the types of core–shell particles can easily be seen by naked eye.
Fig. 2a demonstrates the emission spectrum of LaVO4:Tm3+/Tb3+/Eu3+ (Type 1) core–shell nanoparticles dispersed in dichloromethane (λex = 280 nm). It is evident from the emission spectrum that the peaks corresponding to Tm3+, Tb3+, and Eu3+ emissions can easily be seen. The major emission peak at 475 nm is from 1G4 to 3H6 level of Tm3+ ions, and the one at 545 nm is from 5D4 to 7F5 level of Tb3+ ions. The peaks at 591, 612, 650, and 695 nm are from 5D0 to 7F1, 7F2, 7F3, and 7F4 levels of Eu3+ ions, respectively. The emission peak from Tb3+ ions (490 nm, 5D4 to 7F5) is overlapped with the 475 nm emission of Tm3+ ions. The emission peaks at 575 (5D4 to 7F4) and 625 nm (5D4 to 7F3) from Tb3+ ions also overlapped with Eu3+ ion emissions. The emission spectrum from the type 2 core–shell nanoparticles is shown in Fig. 2b. The emission peaks at 475 nm and 575 nm are from Tm3+ and Dy3+ (4F9/2 to 6H13/2) ions, respectively. The CIE co-ordinates of the emitted light from type 1 and 2 nanoparticles were found to be (0.35, 0.34) and (0.32, 0.34) respectively, compared to (0.33, 0.33) for pure white light (Fig. 2d). Excitation with a range of UV wavelengths (260–330 nm) has also produced bright white light and CIE colour co-ordinates were within the white light region (data not given). In order to prove the non-existence of internal energy transfer between lanthanide ions, both the core–shell type nanoparticles were excited (direct excitation of lanthanide ions) with 410 nm for Eu3+ ions (5D3), 488 nm for Tb3+ ions (5D4), 475 nm for Tm3+ ions (1G4), and 430 nm for Dy3+ ions (4G11/2) light and the emissions corresponding to the direct excitation of lanthanide ions were only observed. This clearly suggests that there was no internal energy transfer between lanthanide-ions suggesting the formation of a core–shell structure with inert LaVO4 shells in between the emissive layers. Our control experiment (doping all the lanthanide ions in the LaVO4 core nanoparticles) did not emit white light (Fig. 3). The emission spectrum (Fig. 3a) shows the presence of Tm3+, Tb3+, and Eu3+ ions emissions, however, the Eu3+ emission dominates compared to Tm3+ and Tb3+ emissions. Similarly, in Fig. 3b, the Dy3+ emission dominates compared to Tm3+ emission. We attribute this to the efficient energy transfer from [VO4]3− group to Eu3+ and Dy3+ ion compared to Tm3+ and Tb3+ ion. This clearly proves the need for a core–shell structure with the inert shell in between the emissive layers.
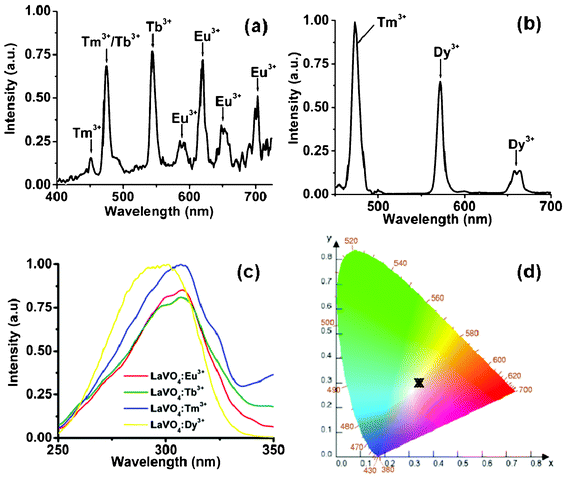 | ||
| Fig. 2 Emission spectra a) LaVO4:Tm3+/Tb3+/Eu3+ (Type 1), b) LaVO4:Tm3+/Dy3+ (Type 2) core–shell nanoparticles (λex = 280 nm), c) excitation spectra of Tm3+, Tb3+, Eu3+, and Dy3+-doped LaVO4 nanoparticles, and d) CIE colour co-ordinates of the white light generated from type 1 and type 2 nanoparticles. | ||
 | ||
| Fig. 3 Emission spectra a) LaVO4:Tm3+/Tb3+/Eu3+ core nanoparticles (control experiment for type 1, λex = 280 nm) and b) LaVO4:Tm3+/Dy3+ core nanoparticles (control experiment for type 2, λex = 280 nm). | ||
Fig. 2c shows the excitation spectra of Tm3+, Tb3+, Dy3+, and Eu3+ ions doped in LaVO4 core nanoparticles. The broad band from 260 to 340 nm clearly proves that the emission from all the Ln3+ ions is mainly due to energy transfer from [VO4]3− group, by a charge-transfer transition in the V–O bond. We note that the excitation peaks of Eu3+, Tb3+, Tm3+, and Dy3+ ions have not been observed due to a dilution effect and efficient energy transfer. This also proves that the lanthanide ions can be excited through energy transfer by using a single UV excitation source with a broad range of wavelength. We have also measured the absolute quantum yield of generated white light as 5 ± 1% and 7 ± 1% for type 1 and type 2 nanoparticles, respectively by using an integrating sphere. The quantum yield of the generated white light is on the lower side compared to bulk material which may be due to the size effect and fluorescence quenching by water molecules involved in the synthesis procedure. We also note that the quantum yield of LaVO4:Eu3+ (5%) core–shell nanoparticles is estimated to be ~75%. It is well known that Tm3+ ion is a weak emitter and its quantum yield is very low (~0.008%). Since, Eu3+ (red) and Dy3+ (yellow) ions are strong emitters, we have reduced their doping concentration to ~0.5% which also limits our white light quantum yield. Furthermore, the average lifetimes of Tm3+, Tb3+, Eu3+, and Dy3+ ions are 0.45 ± 0.05, 1.4 ± 0.1, 1.6 ± 0.12, and 1.1 ± 0.15 ms respectively. (Fig. S1, ESI†) The X-ray powder diffraction pattern (Fig. 4d) of LaVO4 was refined in space group D194h with the Rietveld refinement method and it shows that LaVO4 core–shell nanoparticles are in the highly crystalline zircon type phase in contrast to the bulk monazite phase. This matches with the previous reports.18 Furthermore, FTIR (Fig. S2, ESI†) and 1H NMR (Fig. S3, ESI†) spectra prove the formation of LaVO4 nanoparticles with oleic acid as the stabilizing ligand. Broad bands at 789 and 3000 cm−1 in FTIR show the characteristic peak of the [VO4]3− and the CH stretching vibrations of oleic acid respectively (Fig. S2, ESI†). A broad band above 3000 cm−1 suggests that the surfaces of LaVO4 nanoparticles are terminated with V–OH bonds. Additionally, a broad NMR peak at 5.2 ppm, attributed to the CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH protons clearly suggests the presence of oleic acid (Fig. S3, ESI†).
CH protons clearly suggests the presence of oleic acid (Fig. S3, ESI†).
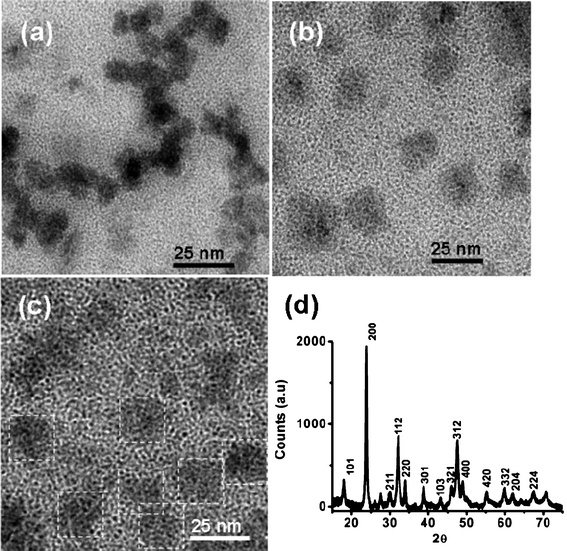 | ||
| Fig. 4 a) TEM image of LaVO4:Eu3+ core nanoparticles, b) TEM image of LaVO4:Tm3+/Tb3+/Eu3+ core–shell nanoparticles, c) TEM image of LaVO4:Tm3+/Dy3+ core–shell nanoparticles (particles are identified with white dotted lines for clarity), and d) XRD pattern of LaVO4:Tm3+/Tb3+/Eu3+ core–shell nanoparticles. | ||
In order to prove the core–shell structure, LaVO4:Eu3+ core, type 1, and type 2 LaVO4 core–shell particles were investigated by point energy dispersive X-ray spectroscopy (EDX), X-ray fluorescence spectroscopy (XRF), fluorescence spectroscopy along with transmission electron microscopy. The TEM images clearly show that the average size of the LaVO4:Eu3+ core (Fig. 4a), type 1 core–shell (Fig. 4b) and type 2 core–shell (Fig. 4c) nanoparticles are 13 ± 2, 19 ± 2, and 23 ± 2 nm, respectively. The TEM images suggest that there is a clear shift in particle size distribution for core–shell particles (average shell thickness of 1 nm per each shell) compared to core particles which clearly suggests the formation of core–shell structure. We note that since the core and shells are made of the same host materials TEM images will not show any contrast between the core and shell structures. In order to further support the formation of the core–shell structure, EDX (Fig. S4 and S5, ESI†) and XRF (Fig. S6 and S7, ESI†) studies have been performed at various points and clearly show the presence of all the lanthanide ions along with carbon and oxygen (due to the presence of oleic acid) in the same composition in various places. This further supports the formation of the core–shell structure. Additionally, increase in the lifetime of Eu3+ ions in the LaVO4 core–shell particles (τav = 1.6 ± 0.12 ms) compared to core particles (τav = 0.98 ± 0.09 ms) suggests that the europium ions are in the core–shell structure and are protected from the external quenching events. This matches with the previous reports and further supports the formation of a core–shell structure. From the above observations, we can eliminate the doubt of formation of separate LaVO4 nanoparticles doped with different lanthanide ions. Moreover, the direct excitation of lanthanide ions (λex = 395 nm for Eu3+, λex = 488 nm for Tb3+, λex = 475 nm for Tm3+ nm for type 1 core–shell particles, and λex = 355 nm for Tm3+ nm for type 2 core–shell particles, and λex = 428 nm for Dy3+ nm) gives only the corresponding lanthanide ion emissions. This clearly proves that there is no internal energy transfer between the lanthanide ions, which also suggests that the shells could be homogeneous.
Conclusions
In conclusion, bright white light has been generated from LaVO4:Tm3+/Tb3+/Eu3+ (Type 1) and LaVO4:Tm3+/Dy3+ (Type 2) core–shell nanoparticles by excitation with a single 280 nm light. The absolute quantum yields of the white light have been found to be 5% and 7% for type 1 and type 2 nanoparticles respectively. We have also shown that emission from all the Ln3+ ions is mainly due to energy transfer from VO4 group and the inert LaVO4 shell between the emissive layers prevents the quenching due to internal energy transfer between lanthanide-ions.Acknowledgements
DST and DST nanomission, India supports are gratefully acknowledged.References
- W. S. Song, K. H. Lee, Y. S. Kim and H. Yang, Mater. Chem. Phys., 2012, 135, 51 CrossRef CAS.
- S. K. K. Shaat, H. C. Swart and O. M. Ntwaeaborwa, Opt. Mater. Express, 2012, 2, 962 CrossRef CAS.
- R. J. R. Vieira, L. Gomes, J. R. Martinelli and N. U. Wetter, Opt. Express, 2012, 20, 12487 CrossRef CAS.
- C. F. Chignell, R. H. Sik and P. J. Bilski, Photochem. Photobiol., 2008, 84, 1291 CrossRef CAS.
- T. Sakamoto, S. Kousaka, K. Uematsu, T. Ishigaki, K. Toda and M. Sato, Phys. Status Solidi C, 2011, 8, 2731 CrossRef CAS.
- G. S. C. Yuen, A. B. Sproul and S. J. Dain, Clin. Exp. Optom., 2010, 93, 66 CrossRef.
- B. Zhang, G. Tan, C. S. Lam, B. Yao, C. L. Ho, L. Liu, Z. Xie, W. Y. Wong, J. Ding and L. Wang, Adv. Mater., 2012, 24, 1873 CrossRef CAS.
- S. Reineke, F. Lindner, G. Schwartz, N. Seidler, K. Walzer, B. Lussem and K. Leo, Nature, 2009, 459, 234 CrossRef CAS.
- X. Zhang, Z. Lu, F. Meng, L. Hu, X. Xu, J. Lin and C. Tang, Mater. Lett., 2012, 79, 292 CrossRef CAS.
- R. Wei, H. Zhang, F. Li and H. Guo, J. Am. Ceram. Soc., 2012, 95, 34 CrossRef CAS.
- W. R. Liu, C. W. Yeh, C. H. Huang, C. C. Lin, Y. C. Chiu, Y. T. Yeh and R. S. Liu, J. Mater. Chem., 2011, 21, 3740 RSC.
- M. J. Vengala Rao Bandi, J. Jeong, K. Jang, H. S. Lee, S. S. Yi and J. H. Jeong, J. Phys. D: Appl. Phys., 2010, 43, 1 Search PubMed.
- G. Li, D. Geng, M. Shang, Y. Zhang, C. Peng, Z. Cheng and J. Lin, J. Phys. Chem. C, 2011, 115, 21882 CAS.
- S. Sivakumar, F. C. J. M. van Veggel and M. Raudsepp, J. Am. Chem. Soc., 2005, 127, 12464 CrossRef CAS.
- G. A. Sotiriou, M. Schneider and S. E. Pratsinis, J. Phys. Chem. C, 2012, 116, 4493 CAS.
- A. Podhorodecki, M. Banski, J. Misiewicz, M. Afzaal, P. O'Brien, D. Cha and X. Wang, J. Mater. Chem., 2012, 22, 5356 RSC.
- P. R. Selvin, Annu. Rev. Biophys. Biomol. Struct., 2002, 31, 275 CrossRef CAS.
- J. H. Chung, J. H. Ryu, S. W. Mhin, K. M. Kim and K. B. Shim, J. Mater. Chem., 2012, 22, 3997 RSC.
- A. Patra, C. S. Friend, R. Kapoor and P. N. Prasad, J. Phys. Chem. B, 2002, 106, 1909 CrossRef CAS.
- A. Patra, S. Saha, M. A. R. C. Alencar, N. Rakov and G. S. Maciel, Chem. Phys. Lett., 2005, 407, 477 CrossRef CAS.
- A. Kar and A. Patra, Nanoscale, 2012, 4, 3608 RSC.
- B. G. You, X. T. Wei, Y. H. Chen, M. Yin and C. K. Duan, J. Lumin., 2012, 132, 2433 CrossRef CAS.
- P. Vergeer, T. J. H. Vlugt, M. H. F. Kox, M. I. den Hertog, J. P. J. M. van der Eerden and A. Meijerink, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 014119 CrossRef.
- V. Mahalingam, F. Mangiarini, F. Vetrone, V. Venkatramu, M. Bettinelli, A. Speghini and J. A. Capobianco, J. Phys. Chem. C, 2008, 112, 17745 CAS.
- M. N. Luwang, R. S. Ningthoujam, S. K. Srivastava and R. K. Vatsa, J. Mater. Chem., 2011, 21, 5326 RSC.
- G. Zhu, Y. Wang, Z. Ci, B. Liu, Y. Shi and S. Xin, J. Lumin., 2012, 132, 531 CrossRef CAS.
- E. Pavitra, G. S. R. Raju, Y. H. Ko and J. S. Yu, Phys. Chem. Chem. Phys., 2012, 14, 11296 RSC.
- S. Dang, J. H. Zhang and Z. M. Sun, J. Mater. Chem., 2012, 22, 8868 RSC.
- S. Sivakumar, J. C. Boyer, E. Bovero and F. C. J. M. van Veggel, J. Mater. Chem., 2009, 19, 2392 RSC.
- A. Patra, C. S. Friend, R. Kapoor and P. N. Prasad, Chem. Mater., 2003, 15, 3650 CrossRef CAS.
- C. Lin, M. T. Berry, R. Anderson, S. Smith and P. S. May, Chem. Mater., 2009, 21, 3406 CrossRef CAS.
- J. C. Boyer and F. C. J. M. van Veggel, Nanoscale, 2010, 2, 1417 RSC.
- E. van der Kolk, P. Dorenbos, K. Krämer, D. Biner and H. U. Güdel, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 125110 CrossRef.
- A. M. Cross, P. S. May, F. C. J. M. v. Veggel and M. T. Berry, J. Phys. Chem. C, 2010, 114, 14740 CAS.
- F. He, P. Yang, D. Wang, N. Niu, S. Gai, X. Li and M. Zhang, Dalton Trans., 2011, 40, 11023 RSC.
- J. W. Stouwdam, M. Raudsepp and F. C. J. M. van Veggel, Langmuir, 2005, 21, 7003 CrossRef CAS.
- J. Liu and Y. D. Li, Adv. Mater., 2007, 19, 1118 CrossRef CAS.
- W. Fan, Y. Bu, X. Song, S. Sun and X. Zhao, Cryst. Growth Des., 2007, 7, 2361 CAS.
- H. Guo, F. Li, R. Wei, H. Zhang and C. Ma, J. Am. Ceram. Soc., 2012, 95, 1178 CrossRef CAS.
- J. W. Stouwdam and F. C. J. M. van Veggel, ChemPhysChem, 2004, 5, 743 CrossRef CAS.
- F. Wang, X. Xue and X. Liu, Angew. Chem., Int. Ed., 2008, 47, 906 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: Decay curve, NMR spectra, and FTIR spectra of LaVO4 samples. See DOI: 10.1039/c2ra21586a |
| This journal is © The Royal Society of Chemistry 2012 |