Efficient purification of chromatin architectural proteins: histones, HMGB proteins and FKBP3 (FKBP25) immunophilin†
Larus E.
Foulger
a,
Connie Goh Then
Sin
b,
Q. Q.
Zhuang
a,
Hugh
Smallman
a,
James M.
Nicholson
c,
Stanley J.
Lambert‡
a,
Colin D.
Reynolds
a,
Mark J.
Dickman
d,
Christopher M.
Wood
*a,
John P.
Baldwin
a and
Katie
Evans
a
aSchool of Pharmacy and Biomolecular Sciences, Liverpool John Moores University, Liverpool, L3 3AF, UK. E-mail: c.m.wood@ljmu.ac.uk, k.evans@ljmu.ac.uk; Fax: +44 (0) 151 231 2170; Tel: +44 (0) 151 231 2334
bSchool of Life and Health Sciences, Aston University, Birmingham, B4 7ET, UK. E-mail: sincgt@aston.ac.uk; Fax: +44 (0)121 204 3696; Tel: +44 (0) 121 204 3467
cDepartment of Macromolecular Crystallography, Diamond Light Source Ltd, Harwell Science and Innovation Campus, Didcot, OX11 ODE, UK. E-mail: james.nicholson@diamond.ac.uk; Fax: 01235 778499; Tel: +44 (0) 1235 778667
dDepartment of Chemical and Biological Engineering, ChELSI Institute, University of Sheffield, Sheffield, S1 3JD, UK. E-mail: m.dickman@sheffield.ac.uk; Fax: +44 (0)114 222 7501; Tel: +44 (0)114 222 7541
First published on 7th September 2012
Abstract
A two-step process of high ionic strength lysis of chicken erythrocyte cell nuclei followed by cation-exchange chromatography has separated at very high yield all the histone and HMGB (high-mobility group B) nuclear proteins, except the less-soluble histone tetramers. Surprisingly high yields of the nuclear immunophilin FKBP3 (FKBP25) and Hsp70 (heat-shock protein 70) co-fractionate with HMGB1 and HMGB3. Furthermore, these proteins can be separated by anion-exchange chromatography. The purified nuclear proteins retain their native, post-translational modification (PTM) marks, including those associated with chromatin-fibre remodelling. These marks are intimately associated with the control of the cell cycle. The methods herein are therefore of value for targeting these and other nuclear proteins for future proteomic studies in healthy and diseased cells.
Introduction
DNA in the nuclei of eukaryotic cells is packaged with histone proteins in nucleosome core particles, where the DNA winds around an octamer of eight core histone proteins in the form of a tetramer and two dimers: ((H3-H4)2-2(H2A-H2B)). Each nucleosome core particle is connected to the next by linker DNA in association with the H1 linker-histone protein. The alternately-concatenated nucleosomes form a chain called a chromatin fibre and this folds into several orders of structure to form the chromosomes of mitotic and interphase nuclei.1 There is a family of non-histone proteins known as the high-mobility group (HMG) that have the ability to bind to nucleosomes and DNA. The HMG proteins fall into three different groups known as HMGB, HMGN and HMGA. Histones, HMG proteins and their associated enzymes that influence DNA binding are called chromatin architectural proteins. Together these proteins protect DNA from unwanted interactions and allow highly selective and timely access to the DNA sequence from factors that control various processes of the cell cycle.HMGB Proteins
The HMGB1, HMGB2 and HMGB3 proteins are ubiquitously expressed and in humans contain two HMG-box domains (A and B) that are DNA-binding and have an ability to bend straight DNA; there is a binding preference for bent DNA.2 There is a long acidic C-terminal domain, whilst the two HMG-box domains are largely positively charged. Each of these domains is formed from three α–helices that form a twisted L shape.2 The chicken HMGB proteins have a high sequence similarity to those of their highly-conserved mammalian counterparts: HMGB1 has 94% of its residues the same as in the human form; HMGB2 has 93% coverage whereas HMGB3 is the lowest at 89%.Though HMGB1 is associated with DNA repair3 and V(D)J recombination,4 this small protein receives the most interest due to the fact that it is also a pro-inflammatory cytokine.5,6 The cytokine activity of HMGB1 lies in its B-box domain7 and antibodies raised against that domain inhibit the cytokine activity of HMGB17in vitro. HMGB1 is released by necrotic cells in large amounts upon trauma and in apoptotic cells it is released by macrophages, neutrophils and monocytes, activated by compounds such as TNF.8–10 Concurrent to the interest of HMGB1 as a drug intervention target is its use as a powerful diagnostic and prognostic tool.11–13
HMGB1 has recently been shown to modulate the degree of activity of telomerase in mammalian cells.14 Inhibition of HMGB1 expression in mouse embryonic fibroblasts caused a reduction in the length of telomeres. Interestingly, prevention of HMGB2 expression had the opposite effect. This difference in function between HMGB1 and HMGB2 is juxtaposed to the similarity of the two sequences. From a structural viewpoint, the large differences in function arise from subtle sequence variations. For example, the amino-acid residues immediately either side of the A-box in HMGB1 are slightly different to the residues in the same locations in HMGB2. This means there would be a difference in the selectivity between the two proteins. Also of interest is the fact that HMGB1 requires a primary binding partner before it can influence telomerase in vitro.
Like HMGB1, HMGB2 is expressed in all cell types. HMGB2 inhibits chondrocyte cell differentiation.15 HMGB2 suppresses pathologic cell growth, unlike HMGB1,16 and regulates cardiac hypertrophy. HMGB2 over-expression has been shown to correlate with poor prognosis in hepatocellular carcinoma.17 However, over-expression of HMGB2 in HPV-positive HeLa cells seems to inhibit progression of cancer cells.18 This discrepancy probably arises because the outcome depends on whether HMGB2 associates with an oncogene or tumour suppressor. It is not just cancer that HMGB2 can affect. It has been implicated in the sex cycle of the Plasmodium parasites, which are involved in the transmission of malaria from mosquitoes to mammals.19 Like HMGB1, HMGB2 also associates with RAGE.20
HMGB3 also contains two HMG box domains. Because of this similar structure, HMGB3, like the other two HMGBs, will associate with either oncogenes or tumor suppressors. It has been implicated in leukaemia when complexed with the oncogene NUP98.21 HMGB3 must be down-regulated in order for myeloid and B-cell differentiation to occur.22–24 HMGB4 is expressed in male germ cells and is not considered herein.25,26
Immunophilins
FK506 binding protein 3 (designated as FKBP3 and still frequently referred to by its former title of FKBP25) is an immunophilin that has a conserved peptidyl-prolyl isomerase (PPI) domain in the C-terminal region that can bind immuno-suppressant drugs. The immunophilins have been divided into three groups. The first group involves the FK506-binding proteins, which bind rapamycin and FK506. The second group is the cyclophilins, which bind cyclosporin.27 Parvulins comprise the third group.28FKBP3 from chicken has a 75% sequence similarity with that of human, a pI of 9.2, and a molecular weight of 25.03 kDa – which is comparable to the molecular weight of HMGB1. The N-terminal domain contains the largest number of differences in amino-acid residues. There are three partial structures of FKBP3 in the Protein Data Bank (PDB): 1PBK, 2KFV and 3KZ7.
It has been shown that FKBP3 functionally associates with histone deacetylases HDAC1 and 2 in HeLa cells and with transcription factor YY1.29 Similarly, it has been shown that porcine HMGB2 is associated with unmodified FKBP3,30 but the evidence for this as a direct interaction in vivo has yet to be established. FKBP3 is localised in the nucleus and associates with nucleolin and casein kinase II.31
A putative model for YY1 transcription-factor suppression
It is the N-terminal region of FKBP3, which is rich in basic amino-acid residues, that interacts with the YY1 transcription factor – not the C-terminal region.32 This interaction would increase the binding of YY1 to DNA enhancing the former's repression characteristics. There is other evidence suggesting that FKBP3 acts as a partner for MDM2, a factor involved in P53 regulation, and may act as a histone chaperone.The fundamental interactions between YY1, HDACs 1 and 2 and FKBP3 have been stressed,32,33 clearly indicating a role for FKBP3 in the repression activity of YY1. The latter transcription factor is a zinc-finger DNA-binding protein. It has been shown that in nuclear extracts from a virally transformed chicken-erythroid cell line there is an interaction with DNA in a mechanism leading to the repression of the zinc-finger hematopoietic transcription factor GATA1.34
These studies therefore identify molecular assemblies that are reputed to control the repression properties of YY1. The proteins from these assemblies can be extracted from tissue samples and the gentle extraction techniques used herein allow that goal to be achieved. In addition, the components of any assembly will retain the post-translational modifications they had in vivo. The targeting of these proteins makes the methods our group have developed useful in the expanding field of clinical epigenetic analysis.
Fractionation of native nuclear proteins
The original method for large-scale fractionation of HMGB proteins used calf-thymus chromatin that was extracted three times in 0.35 M NaCl, pH 7, combining the supernatants after a 2000 g centrifugation.35 The extracts were made 2% in TCA (trichloro acetic acid), passed through sintered glass filters and precipitated in three volumes of acidified acetone. CM-Sephadex C25 ion-exchange fractionation was used to purify the HMGs.Later it was shown that both histones and non-histone proteins from chicken-erythrocyte nuclei could be dissociated in 2 M NaCl, 10 mM Tris, pH 7.5.36 They were fractionated on a P11 phospho-cellulose column, eluting them with 0–2 M NaCl, pH 9.0. There was separation between core histones, linker histone H1, HMGs and linker histone H5.
The purification of core histones as octamers in 2 M NaCl showed the usefulness of using high-salt conditions.37 Crystallisation of the core histones in ammonium sulphate established the nature of the histone-fold-pair structures between H2A and H2B and between H3 and H4.38
More recently, a technology for purifying nuclear proteins in various strict conditions in KCl/phosphate solutions was developed in our laboratory. This led to high-resolution structures of the histone octamer39,40 and details of the post-translational modifications of chicken-erythrocyte linker histones.41 Herein described is a modification of this technology that has led to a procedure for preparing HMGB1, 2 and 3, linker histones, histone dimers and FKBP3 from chicken erythrocytes. It was found that FKBP3 fractionated at the same time as HMGB1 and 3 using cation-exchange chromatography at a pH of around 5.7.
Chicken-erythrocyte nuclear proteins were chosen for the present work for three reasons. Firstly, chicken erythrocytes are terminally differentiated, with only globin genes active plus a few house-keeping genes.42 Secondly, large quantities of chicken-erythrocyte cell nuclei (5 × 1012 yield) can be quickly prepared in 10 mM NaCl, 10 mM Tris, pH 7.5 with protease inhibitors, lysing cells in 1% Triton™ X100, which is based on a procedure from 1975.43 Thirdly, native HMG proteins are traditionally extracted from cell nuclei using 0.35 M NaCl and then dissolved in highly acidic conditions. However a large yield of HMG-rich, (but histone-depleted) proteins from intact chicken-erythrocyte nuclei was extracted in 80 mM KCl, 80 mM equimolar phosphate in our laboratory.44 In addition, there were around 500 other proteins that were identified by mass spectrometry. This batch of proteins were in a supernatant obtained after pelleting the intact nuclei. This supernatant included all the proteins in the putative YY1 repression model, including YY1, GATA1, HDACs, FKBP3, HMGs and H2A.Z as a moiety.
Cation-exchange chromatography of a chicken-erythrocyte nuclei lysate
Our group developed a simple technique to maximise the yield of proteins from the nucleus during lysis. The key is to expose all nuclei to the lysis solution. Failure to do this leads to dissociated histones in the core-histone octamer.In this technique 80 mL of chicken-erythrocyte cell nuclei (2 × 109 ml−1) were directly lysed in ultra-centrifugation tubes by layering 1 volume of nuclei in each tube onto 4 volumes of lysis buffer, 2 M KCl, 0.775 M K2HPO4, 0.775 M KH2PO4, 2.5 mM benzamidine hydrochloride (as a protease inhibitor). Each tube was suddenly shaken so that the slight delay in nuclei lysis gave individual cell lysis under uniform solvent conditions. The gel lysate was ultra-centrifuged at 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g for eighteen hours and the DNA-free supernatant was dialysed into buffer A (100 mM KCl, 100 mM KH2PO4, pH 4.5). The supernatant was concentrated to 40 mls for cation-exchange chromatography using four Hi-Trap, fast-flow SP Sepharose columns.
000 g for eighteen hours and the DNA-free supernatant was dialysed into buffer A (100 mM KCl, 100 mM KH2PO4, pH 4.5). The supernatant was concentrated to 40 mls for cation-exchange chromatography using four Hi-Trap, fast-flow SP Sepharose columns.
Linear cation-exchange chromatography was carried out in an AKTA liquid chromatography system, with buffer B (2 M KCl, 100 mM K2HPO4, pH 9) being used to elute adsorbed proteins. Fig. 1 is the chromatogram showing essentially seven resolved maxima. SDS poly-acrylamide gel electrophoresis (SDS PAGE) of each peak is shown in Fig. 2a and 2b.
 | ||
| Fig. 1 Cation-exchange chromatography of proteins in the chicken erythrocyte lysate. Four Hi-Trap, fast-flow SP Sepharose columns were used. Buffer A was 100 mM KCl, 100 mM KH2PO4, pH 4.5 and buffer B was 2 M KCl, 100 mM K2HPO4, pH 9. 10 mls of 15 mg ml−1 protein sample was loaded onto the column and was run at a rate of 5 mL min−1 for 60 column volumes (1200 mL in total). | ||
 | ||
| Fig. 2 a) 20% SDS PAGE of the cation-exchange chromatography peaks of Fig. 1, stained in colloidal Coomassie brilliant blue: heavy loading. The object of overloading the gel was to generate bands of any very-low abundance proteins. This was particularly effective in revealing minority proteins in peak 7. The bands in the lanes were cut out for analysis by mass spectrometry. CYP B is an abbreviation of cyclophilin B. b) Second 20% SDS PAGE of the cation-exchange chromatography peaks of Fig. 1, lighter loading. | ||
The protein in the total lysate was estimated by UV spectrophotometry to be approximately 100 mg. Proteins in the peaks were identified using tandem mass spectrometry analysis in conjunction with in-gel tryptic digestion of the corresponding bands (see ESI†) cut from the SDS PAGE gels. The yield of each band was estimated very approximately from the areas under peaks in the chromatograms.
In summary, cation-exchange chromatography of a high ionic strength lysate of chicken erythrocyte cell nuclei is sufficient to purify, in one step, and with high yield, several of the chromatin architectural proteins. The contents of peaks 1 to 7 in Fig. 1 and 2 can be collated using the data in the ESI.†
Peak 1 contains intense bands of HMGB1 and HMGB3, significant bands of immunophilins FKBP3 and cyclophilin B and small quantities of endothelial differentiation-related factor 1. Fig. 7 (see later) shows quite strong higher-molecular weight proteins are also present. The most notable are HSP70 and proliferation-associated protein 1.
Peak 2 contains a lower yield of total protein compared with peak 1, including a very intense, dominating band of HMGB2, together with very small quantities of HMGB1 and 3. Also present are linker-histone fragments, cyclophilin B, HMGN, RNA polymerase II and transcription co-factor 4.
Peak 3 contains linker histones H1a and b.
Peak 4 contains linker histone H1b.
Peak 5 contains core-histone dimers.44
Peak 6 contains pure linker histone H5.
Peak 7 contains a very small quantity of core-histone tetramers with H5. It is not yet clear if these two bind together. However, peaks 5 and 7 exactly correspond to the separation of pure histone dimers and pure histone tetramers respectively, as derived from pure histone octamers by cation-exchange chromatography.44
It should be mentioned that the high ionic strength lysis conditions are above the solubility limits for histone octamers, but clearly histone dimers exist on their own under these conditions in the soluble fraction of the lysate. In our laboratory we are also interested in structural studies of the proteins and complexes we produce. Such studies are facilitated by the use of circular dichroism and X-ray crystallography. During on-going experiments to crystallise purified histone tetramers and dimers, it has been noticed that there is a remarkable difference in the solubilities of these two complexes. The dimers are soluble above 2 M KCl, 1.6 M equimolar phosphate, whereas the histone tetramers, (H3-H4)2, precipitate at 1 M KCl, 1 M equimolar phosphate. Furthermore, synchrotron radiation circular dichroism shows that the dimers have the same α-helical content as in the complete octamer whereas the tetramers have lost some helical content (unpublished data).
High-resolution cation-exchange chromatography of the chicken-erythrocyte nuclei lysate
The resolution achievable in a chromatograhy system depends primarily on the efficiency, selectivity and retention capacity of the system. Efficiency is directly proportional to column length. Increasing the efficiency will result in narrower peaks and thus better separation of overlapping peaks. Therefore the strategy used by the authors to increase resolution was to use multiple, concatenated columns. The purpose of improving column resolution was to increase the separation between peaks 1 and 2 in Fig. 1 that contain the HMG proteins.Eight rather than four 5 ml Hi-Trap, fast-flow SP Sepharose columns were therefore used to produce a new chromatogram, in such a way as to collect as many fractions as possible in the region of peaks 1 and 2. The total lysate described in the previous section was dialysed into buffer A again (100 mM KCl, 100 mM KH2PO4) at pH 4.5. Cation-exchange buffer B was used again to elute the proteins but in this case it was altered (1 M KCl, 100 mM K2HPO4). The chromatography column was run at 5 ml min−1 starting at 9% B to 25% B over 30 column volumes, giving an elution volume of 1200 ml.
This time peak 1 is completely resolved from peak 2 in the chromatogram of Fig. 3 and the SDS PAGE gel of Fig. 4 confirms that HMGB1, HMGB3 and FKBP3 fractionate in peak 1 whereas HMGB2 is completely resolved in peak 2. It is clear that eight Hi-Trap, fast-flow SP Sepharose cation-exchange columns can resolve the linker histones in peak 3 and peak 4 lanes of Fig. 2, namely H1a and H1b.
 | ||
| Fig. 3 Cation-exchange chromatography of peaks from the first cation-exchange run. Eight Hi-Trap, fast-flow SP Sepharose columns were used with the soluble proteins from the nuclei lysate to improve the resolution of the cation-exchange chromatography of Fig. 1. As before, buffer A was 100 mM KCl, 100 mM KH2PO4 at pH 4.5 and buffer B was 1 M KCl, 100 mM K2HPO4 to elute the proteins. The chromatogram was run at 5 ml min−1 but just peaks 1 and 2 were fractionated by eluting only over the limiting range from 9% B to 25% B over 30 column volumes. | ||
 | ||
| Fig. 4 A 20% SDS PAGE gel of the high-resolution cation-exchange chromatography peaks of Fig. 3. HMGB1 and HMGB3 occur in peak 1, separated from HMGB2 in peak 2. FKBP3 occurs in lanes peak 1 and peak 2, but is most pronounced in the former. | ||
HMGB1 with HMGB2 have been separated from other chromosomal proteins before45 using a 0.35 M NaCl extraction from steer thymus nuclei and separation on a Mono S® column at pH 6 in 20 mM MES buffer, but the other proteins identified herein were not isolated.
Anion-exchange chromatography of key cation-exchange peaks
Next, four 5 ml DEAE anion-exchange columns (HITrap, fast flow, GE Healthcare) were equilibrated with buffer A: 10 mM KCl, 9.4 mM K2HPO4, 6 mM KH2PO4, pH 8.0. The fractions from peaks 1 and 2 of Fig. 1 were combined and dialysed into buffer A, loaded onto the columns and eluted with buffer B: 2 M KCl, 9.4 mM K2HPO4, 6 mM KH2PO4, pH 8.0.The resulting chromatogram (Fig. 5) shows firstly a non-binding peak that mass spectrometry analysis of SDS PAGE bands (Fig. 6) has shown to contain FKBP3 as well as cyclophilin B. The latter is known to be present in the peak 2 lanes of Fig. 2 and 4, and to a lesser extent in the peak 1 lanes, so an anion-exchange chromatogram of peak 1 of Fig. 4 would produce pure FKBP3.
 | ||
| Fig. 5 Anion-exchange chromatography of cation-exchange peaks 1 and 2. Four 5 ml Hi-Trap, fast-flow DEAE anion-exchange columns were equilibrated with buffer A: 10 mM KCl, 9.4 mM K2HPO4, 6 mM KH2PO4, pH 8.0. The fractions from peaks 1 and 2 of Fig. 1 were combined and dialysed into buffer A, loaded onto the columns and eluted on a linear gradient with buffer B: 2 M KCl, 9.4 mM K2HPO4, 6 mM KH2PO4, pH 8.0. The elution gradient ran from 0 to 58% of buffer B. The column was run for 60 column volumes (1200 ml) at 5 mls min−1. Notice how the FKBP3 is this time washed out, whilst HMGB1 co-elutes with HMGB2. | ||
 | ||
| Fig. 6 A 20% SDS PAGE gel of the peak fractions of Fig. 5. FKBP3 and cyclophilin B form part of the un-bound fraction. Peak 1 contains HMGB3 and peak 2 contains HMGB1 with HMGB2. | ||
The chromatogram of Fig. 5 shows two peaks, the first of which is identified in Fig. 6 to be pure HMGB3 and the second peak to be HMGB1 and HMGB2 with some HMGB3. A comparison between Fig. 4 and 6 therefore shows that HMGB1, HMGB2 and HMGB3 can be individually purified in several milligram quantities by successive fractionation of the lysate, first by cation-exchange chromatography and then by anion-exchange chromatography. Concomitantly, approximately one milligram of FKBP3 and CYPB can be purified.
Discussion
Approximately 100 mg of soluble nuclear proteins in a supernatant were extracted by lysis of pure chicken-erythrocyte cell nuclei in high-KCl/phosphate. The DNA and insoluble biomolecules were centrifuged to a pellet at 100000 g. The result was the fractionation of tens of milligrams of reasonably pure histone dimers (H2A-H2B) and the two linker histones H1 and H5. A combination of cation-exchange and anion-exchange chromatography led to the purification of several milligrams of HMGB1, HMGB2 and HMGB3. Similarly, milligram quantities of the immunophilins FKBP3 and cyclophilin B were produced.
Fractionation of native chromatin-architectural proteins provides opportunities to study their native binding partners inter se and with other nuclear proteins. Purified FKBP3 will be valuable in studying the reputed interactions with HDACs 1 and 2 and YY1, leading to the repression of GATA1 and in determining which proline residues in which proteins become isomerised. GATA1 has a major role in the dominant expression of globin genes in chicken-erythrocytes:42 so this no doubt accounts for the relatively high yield of FKBP3.
The binding of FKBP3 with HMGB2 in porcine brain has been identified.30 The question is whether the HMGBs are also involved in the assemblies involved in YY1 repression. Fig. 2 and 4 indicate that, in peak 1, HMGB1 and/or 3 might bind FKBP3 under our conditions because they fractionate together using cation-exchange chromatography. However there is no FKBP3 in peak 2 with the HMGB2, as might be expected by the results of Leclercq et al.30
The theoretical charges on HMGB1, HMGB2, HMGB3, FKBP3 and FKBP3-HMGB1 at pH 5.5 are 1.8, 10.0, 10.1, 13.1 and 14.9, respectively. The charges were calculated using pI values. FKBP3 has a high pI of 9.2 and HMGB1 has a low pI of 5.7. The pIs of the remaining HMGBs are in between – at about 8.5. The three proteins could therefore fractionate together by cation-exchange chromatography if HMGB1 was coupled to FKBP3. However HMG-binding to cation exchangers is strongly influenced by their unique, completely acidic, C-terminal regions.
Binding between FKBP3 and HMGB proteins could not exist at pH 7.5 to 8.0, since the immunophilins do not bind to the anion-exchange medium in Fig. 5, whereas the HMGBs do. Moreover, experiments at pH 7.5 to crosslink the proteins in peak 1 of Fig. 1 and 2, following closely the procedures of Cato et al.,46 yielded no zero-length crosslinking between HMGs and FKBP3. The latter remained completely unaffected by the crosslinking (data not shown). The crosslinking within the HMGs during this experiment agreed with the results of Cato et al.46 and there was some crosslinking between HMGs and cyclophilin fragments in diffuse higher-molecular-weight bands.
Fig. 7 shows gel-exclusion chromatography of the peak 1 proteins from peak 1 of Fig. 1 and 2 at pH 6.0. The dominant peak in the chromatogram still contains the familiar signature of Fig. 2 and 4, namely FKBP3 below which are HMGB1 and HMGB3. The molecular weights of FKBP3 (25 kDa) and HMGs (24.9 kDa for HMGB1) are very close, which accounts for the single peak in Fig. 7. However there is no peak in an earlier fraction that should arise if there was a complex of FKBP3 with an HMGB protein. Binding between an HMGB protein with FKBP3 therefore cannot be confirmed even at pH 6.0. The large peak in the gel-filtration analysis also contains CAPRIN1 (proliferation-associated protein 1) which has a considerably larger mass than the other constituents of the peak.
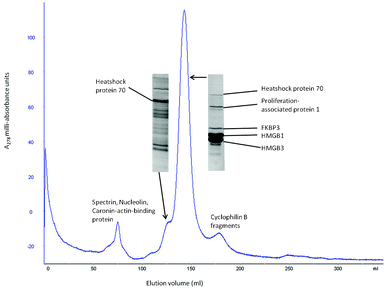 | ||
| Fig. 7 Gel-filtration of peak 1 from Fig. 3. To determine if the FKBP3 in peak 1 of Fig. 3 formed a complex with either HMGB1 or HMGB3, the fractions from that peak were passed through a 0.5 metre long S100 gel-filtration column. The analysis was carried out in buffer at pH 6 (100 mM KCl, 95 mM KH2PO4, 5 mM K2HPO4). There is an early minor peak of spectrin casein-actin-binding protein and nucleolin. Following this is a significant peak rich in Hsp 70 which is close to the main peak of proliferation-associated protein 1, FKBP3, HMGB1 and HMGB3. A later significant peak contained largely different fragments of cyclophilin B. | ||
It is interesting to note that the CAPRIN1 co-fractionates with the FKBP3 and HMGBs (see Fig. 7). Also, the yield of a pronounced peak containing largely heat-shock protein HSP70 is fairly high. Other acidic higher molecular weight proteins nucleolin, spectrin and caronin-actin-binding protein are also in peak 1 of Fig. 2 and 4.
It is essential to know the structures of nuclear protein assemblies that are involved in the cell cycle to the highest resolution, particularly those that relate to the control of transcription. Reconstruction of protein assemblies from recombinant proteins has been very successful. However, this can be improved upon by the fractionation of pure nuclear proteins or assemblies thereof, including native PTM marks, from their cell nuclei.
HMGB proteins are known to bind to many proteins in the cell nucleus26 and operate over many signalling pathways. It follows therefore, that there will be primary binding partners of the HMGB proteins.14 Some of those primary binding partners have been isolated by the work herein, but the methods used can easily be adapted to identify other primary binding partners.
Conclusions
The technology presented herein allows the efficient isolation and high-yield purification of chromatin architectural proteins using gentle procedures that maintain the epigenetic marks on those proteins. There is a growing demand for clinical based epigenetic-mark analysis, and our group is now developing the technology for use on human blood samples.In addition to protein purification, the technology allows the isolation of potential molecular complexes which can be analysed by gel exclusion chromatography and mass spectrometry. Our results so far do not confirm that HMGB1, 2 and 3 bind to FKBP3.
Acknowledgements
We are grateful for the supporting work of previous project and research students, particularly Drs Sirirath Sodngam and Sayampong Pongdam.References
- B. M. Turner, ed Frontmatter, in Chromatin and Gene Regulation: Mechanisms in Epigenetics, 2007, Blackwell Science Ltd, Oxford, UK. DOI:10.1002/9780470750629.fmatter.
- U. Andersson, H. Erlandsson-Harris, H. Yang and K. J. Tracey, J. Leukoc. Biol., 2002, 72, 1084–1091 CAS.
- S. S. Lange, D. L. Mitchel and K. M. Vasquez, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10320–10325 CrossRef CAS.
- P. C. Swanson, Mol. Cell. Biol., 2002, 22, 1340–1321 CrossRef CAS.
- H. Wang, O. Bloom, M. Zhang, J. M. Vishnubhakat, M. Ombrellino, J. Che, A. Frazier, H. Yang, S. Ivanova, L. Borovikova, K. R. Manogue, E. Faist, E. Abraham, J. Andersson, U. Andersson, P. E. Molina, N. N. Abumrad, A. Sama and K. J. Tracey, Science, 1999, 285, 248–251 CrossRef CAS.
- E. Abraham, Crit. Care, 2009, 13, 1004 CrossRef.
- J. Li, R. Kokkola, S. Tabibzadeh, R. Yang, M. Ochani, X. Qiang, H. E. Harris, C. J. Czura, H. Wang, L. Ulloa, H. Wang, H. S. Warren, L. L. Moldawer, M. P. Fink, U. Andersson, K. J. Tracey and H. Yang, Mol. Med., 2003, 9, 37–45 CAS.
- J. Gauley and D. S. Pisetsky, Autoimmunity, 2009, 42, 299–301 CrossRef CAS.
- V. Urbonaviciute, B. G. Fürnrohr, C. Weber, M. Haslbeck, S. Wilhelm, M. Herrmann and R. E. Voll, J. Leukocyte Biol., 2007, 81, 67–74 CrossRef CAS.
- M. J. Cohen, K. Brohi, C. S. Calfee, P. Rahn, B. B. Chesebro, S. C. Christiaans, M. Carles, M. Howard and J. F. Pittet, Crit. Care, 2009, 13, R174 CrossRef.
- M. Rosas-Ballina, R. S. Goldstein, M. Gallowitsch-Puerta, L. Yang, S. I. Valdés-Ferrer, N. B. Patel, S. Chavan, Y. Al-Abed, H. Yang and K. J. Tracey, Mol. Med., 2009, 15, 195–202 CAS.
- S. Zhu, M. Ashok, J. Li, W. Li, H. Yang, P. Wang, K. J. Tracey, A. E. Sama and H. Wang, Mol. Med., 2009, 15, 275–282 CAS.
- S. Gibot, F. Massin, A. Cravoisy, D. Barraud, L. Nace, B. Levy and P. E. Bollaert, Intensive Care Med., 2007, 33, 1347–1353 CrossRef CAS.
- E. Polanská, Z. Dobšáková, M. Dvořáčková, J. Fajkus and M. Stros, Chromosoma, 2012, 121, 419–431 CrossRef.
- N. Taniguchi, B. Caramés, E. Hsu, S. Cherqui, Y. Kawakami and M. Lotz, J. Biol. Chem., 2011, 286, 41489–41498 CrossRef CAS.
- S. Franklin, H. Chen, S. Mitchell-Jordan, S. Ren, Y. Wang and T. M. Vondriska, Mol. Cell. Proteomics, 2012, 11, mcp.M111.014258 Search PubMed.
- J. H. Kwon, J. Kim, J. Y. Park, S. M. Hong, C. W. Park, S. J. Hong, S. Y. Park, Y. J. Choi, I. G. Do, J. W. Joh, D. S. Kim and K. Y. Choi, Clin. Cancer Res., 2010, 16, 5511–5521 CrossRef CAS.
- D. Lee, J. H. Kwon, E. H. Kim, E. S. Kim and K. Y. Choi, Cancer Lett., 2010, 292, 125–132 CrossRef CAS.
- M. Gissot, L. M. Ting, T. M. Daly, L. W. Bergman, P. Sinnis and K. Kim, J. Biol. Chem., 2008, 283, 17030–17038 CrossRef CAS.
- T. Pusterla, F. de Marchis, R. Palumbo and M. E Bianchi, Autoimmunity, 2009, 42, 308–310 CrossRef CAS.
- A. Petit, C. Ragu, V. Della-Valle, M. J. Mozziconacci, M. Lafage-Pochitaloff, G. Soler, C. Schluth, I. Radford, C. Ottolenghi, O. A. Bernard, V. Penard-Lacronique and S. P. Romana, Leukemia, 2010, 24, 654–658 CrossRef CAS.
- M. J. Nemeth, D. J. Curtis, M. R. Kirby, L. J. Garrett-Beal, N. E. Seidel, A. P. Cline and D. M. Bodine, Blood, 2003, 102, 1298–1306 CrossRef CAS.
- M. J. Nemeth, A. P. Cline, S. M. Anderson, L. J. Garrett-Beal and D. M. Bodine, Blood, 2005, 105, 627–634 CrossRef CAS.
- M. J. Nemeth, M. R. Kirby and D. M. Bodine, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13783–13788 CrossRef CAS.
- R. Catena, E. Escoffier, C. Caron, S. Khochbin, I. Martianov and I. Davidson, Biol. Reprod., 2009, 80, 358–66 CrossRef CAS.
- M. Stros, Biochim. Biophys. Acta, Gene Regul. Mech., 2010, 1799, 101–113 CAS.
- R. Miele, M. Borro, M. L. Mangoni, M. Simmaco and D. Barra, Peptides, 2003, 24, 1713–1721 CrossRef CAS.
- P. E. Shaw, EMBO Rep., 2007, 8, 40–45 CrossRef CAS.
- W. M. Yang, Y. L. Yao and E. Seto, EMBO J., 2001, 20, 4814–825 CrossRef CAS.
- M. Leclercq, F. Vinci and A. Galat, Arch. Biochem. Biophys., 2000, 380, 20–28 CrossRef CAS.
- W. M. Yang, Y. L. Yao and E. Seto, EMBO J., 2001, 20, 4814–4825 CrossRef CAS.
- Y. L. Yao, Y. C. Liang, H. H. Huang and W. M. Yang, Curr. Opin. Pharmacol., 2011, 11, 301–307 CrossRef CAS.
- D. Dilworth, G. Gudavicius, A. Leung and C. J. Nelson, Biochem. Cell Biol., 2012, 90, 1–15 Search PubMed.
- H. Rincon-Arano, V. Valadez-Graham, G. Guerrero, M. Escamilla-Del-Arenal and F. Recillas-Targa, J. Mol. Biol., 2005, 349, 961–975 CrossRef CAS.
- G. H. Goodwin, R. H. Nicolas and E. W. Johns, Biochim. Biophys. Acta, Protein Struct., 1975, 405, 280–291 CrossRef CAS.
- L. C. Garg and G. R. Reeck, Protein Expression Purif., 1998, 14, 155–159 CrossRef CAS.
- R. W. Burlingame, W. E. Love, B. C. Wang, R. Hamlin, H. X. Nguyen and E. N. Modrianakis, Science, 1985, 228, 546–543 CAS.
- G. Arents, R. W. Burlingame, B. C. Wang, W. E. Love and E. N. Moudrianakis, Proc. Natl. Acad. Sci. U. S. A., 1991, 15, 10148–10152 CrossRef.
- L. Chantalat, J. M. Nicholson, S. J. Lambert, A. J. Reid, M. J. Donovan, C. D. Reynolds, C. M. Wood and J. P. Baldwin, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2003, 59, 1395–1407 CrossRef CAS.
- C. M. Wood, J. M. Nicholson, S. J. Lambert, L. Chantalat, C. D. Reynolds and J. P. Baldwin, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2005, 61, 541–545 CrossRef.
- A. P. L. Snijders, S. Pongdam, S. J. Lambert, C. M. Wood, J. P. Baldwin and M. J. Dickman, J. Proteome Res., 2008, 7, 4326–4335 CrossRef CAS.
- T. R. Hebbes, A. L. Clayton, A. W. Thorne and C. Crane-Robinson, EMBO J., 1994, 13, 1823–1830 CAS.
- W. T. Garrard and R. Hancock, Methods Cell Biol., 1978, 17, 27–50 CrossRef CAS.
- Q. Zhuang, PhD thesis, Liverool John Moores University, 2012 Search PubMed.
- G. A. Rice and R. D. Cole, Protein Expression Purif., 1990, 1, 87–92 CrossRef CAS.
- L. Cato, K. Stott, M. Watson and J. O. Thomas, J. Mol. Biol., 2008, 384, 1262–1272 CrossRef CAS.
Footnotes |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra21758a |
| ‡ Deceased. We wish to acknowledge, with affection and gratitude, the work of our colleague, the late Dr Stan Lambert. |
| This journal is © The Royal Society of Chemistry 2012 |