Facile synthesis of upconversion luminescent mesoporous Y2O3:Er microspheres and metal enhancement using gold nanoparticles
Peng
Zhao
,
Yihua
Zhu
*,
Xiaoling
Yang
,
Kaicai
Fan
,
Jianhua
Shen
and
Chunzhong
Li
Key Laboratory for Ultrafine Materials of Ministry of Education, School of Materials Science and Engineering, East China University of Science and Technology, Shanghai 200237, China. E-mail: yhzhu@ecust.edu.cn; Fax: +86 21 6425 0624; Tel: +86 21 6425 2022
First published on 7th September 2012
Abstract
Lanthanide-doped upconversion (UC) nanocrystals display the property of emitting visible light following photoexcitation with near-infrared laser light, which has attracted much interest because of its great potential in biological fields. Recently, the coupling of UC nanocrystals with metal nanoparticles (NPs) has been developed as a valuable strategy to enhance their luminescence. Herein, we present a facile method to fabricate mesostructured Y2O3:Er UC microspheres using mesoporous silica spheres as a hard template, and then integrate Y2O3:Er UC microspheres with Au NPs for constructing Y2O3:Er@Au hybrid composites, in which a multilayer polyelectrolyte serves as spacer. We further demonstrate the multicolour UC emissions are enhanced after adsorbing Au NPs and this enhancement can be at least partly attributed to surface plasmon-coupled emission, which can increase the radiative decay rate and emission efficiency. It is anticipated that these hybrid nanostructures may provide a platform for widely exploring applications in bioimaging, bioassays and detection.
Introduction
Upconversion (UC) refers to nonlinear optical processes in which the sequential adsorption of two or more photons leads to the emission of light at shorter wavelength than the excitation wavelength (anti-Stokes emission).1 Compared to quantum dots and conventional organic dye markers, UC nanoparticles (NPs) have several advantages including high chemical stability, low toxicity, and a high signal-to-noise ratio, and are promising luminescent probes for biological species.2–4 However, UC luminescence intensity drops precipitously with excitation power in the low-excitation-power region. A primary obstacle to the incorporation of UC phosphors into real devices and applications has been the inability to obtain high upconversion efficiencies under modest excitation flux.5–7Nanosized noble metal structures exhibit an extinction band in or close to the visible range, which is not present in the bulk noble metal spectrum. This band is attributed to localized surface plasmon resonance (LSPR), which is the collective electron-cloud oscillation on a noble metal surface and is caused by the interaction of the noble metal with incident light.8–11 The local electric fields generated by LSPR in the vicinity of the NPs can significantly modify the spectroscopic properties of neighboring fluorophores and cause an enhancement or quenching of the photoluminescence (PL) relative to the native state, which depends on the distance between the metal surface and the material.11–16 The distance dependence of PL enhancement or quenching has so far been observed with organic dyes,17 quantum dots,18,19 and rare-earth complexes20 with different materials as spacers. Potential applications of metal-induced PL enhancement or quenching range from sensing technologies21 to solid-state lighting.22
A great deal of research effort has been focused on the incorporation of noble metal NPs on NaYF4:Ln (Ln = Yb, Er, Ho),23–28 however, the integration of Y2O3:Ln with plasmonic nanocrystals remains largely unexplored. Besides, there are few reports on the controlled synthesis of Y2O3 microstructures.29 As Y2O3 is an attractive host for phosphors activated with lanthanide ions, it is reasonable to expect that the integration of mesostructured Y2O3 with plasmonic nanocrystals will play an important role in biological imaging, therapeutics and catalysis.30
Herein, we present a rational route to design and synthesize mesostructured Y2O3:Er UC microspheres and combine Au NPs using a multilayer polyelectrolyte as an interface for Au decoration (Fig. 1). With the mesopores serving as confined reactors or growing spaces, mesoporous silica (MS) spheres were used as a hard template to fabricate mesostructured Y2O3:Er UC microspheres. Specially, a ligand-exchange process was carried out by using poly(allylamine hydrochloride) (PAH) and poly(sodium-p-styrenesulfonate) (PSS) as multidentate ligands that displace the original hydrophobic ligands on the UC NPs. We used the polyelectrolyte layer-by-layer (LbL) approach, involving alternate binding of oppositely charged polyelectrolyte layers via electrostatic interactions. Finally, negatively charged Au NPs were prepared separately and attached to the microspheres. On the other hand, the Er3+ doped mesostructured Y2O3:Er microspheres showed UC luminescence through single excitation at 980 nm. The power dependence of UC emission intensities under the excitation of 980 nm was investigated. It was interesting to note that the attachment of Au NPs can enhance the UC emission.
 | ||
| Fig. 1 Schematic diagram and structural models of the mesostructured Y2O3:Er@Au. | ||
Experimental section
Reagents and materials
PAH and PSS were purchased from Sigma-Aldrich Chemicals Co. Y(NO)3 (99.9%) and Er(NO)3 (99.9%) were obtained from Alfa Aesar Co. Ltd. All other chemicals were purchased from the Shanghai Chemical Reagent Co. All chemicals were used as received. Ultrapure water (18 MΩ cm) was used for all experiments.Synthesis of mesostructured Y2O3:Er microspheres
MS spheres were synthesized as described in the literature31,32 with a little modification. Briefly, 1 g of hexadecylamine was dissolved in a mixed solution including 90 mL ultrapure water and 100 mL isopropanol. Then 1.4 mL ammonia was added, which was followed by the addition of 5.8 mL of tetraethoxysilane. The mixture was homogenized and kept at room temperature overnight. The resulting white precipitate was collected by filtration of the reaction mixture, washed with ethanol and dried at room temperature. The templates were removed by calcination at 600 °C for 6 h. To expand the pore size of the MS spheres, a mixed solution with three ingredients including NaCl, LiCl and KNO3 was adopted. MS spheres were soaked in the above salt solution and dispersed by ultrasound. The mixture was dried at 80 °C, and then calcined at 400 °C for 2 h.For a typical synthesis of mesostructured Y2O3:Er (5 wt%) microspheres, briefly, 1 g of MS sphere powder, 0.95 mL 1.0 M Y(NO3)3, 50 μL 1 M Er(NO3)3 and 6 g urea were mixed with 500 mL H2O with continuous stirring overnight. Then the mixture was heated at 90 °C for 2 h. The resulting dispersions were centrifuged and resuspended with ethanol several times, and the product was dried and stored in a desiccator overnight to obtain the MS/(Y, Er)(OH)CO3·H2O microspheres.15
| H2N–CO–NH2 → NH4+ + OCN− | (1) |
| OCN− + OH− + H2O → NH3 + CO32− | (2) |
| (Y, Er)OH(H2O)n2+ + CO2 + H2O → (Y, Er)OHCO3·H2O + 2H+ + (n − 1)H2O | (3) |
The MS/Y2O3:Er microspheres were obtained by thermal treatment of the precursor at 700 °C for 3 h (2 °C min−1 to 700 °C); the morphology was preserved after the heat treatment. The mesostructured Y2O3:Er microspheres were obtained by removing MS spheres using NaOH.
LbL assembly of polyelectrolyte
The LbL procedure was carried out using the positive polyelectrolyte PAH and the negative polyelectrolyte PSS. Briefly, the as-prepared mesostructured Y2O3:Er microspheres were dispersed in 6 mL of PAH (1 g L−1) containing 0.5 M NaCl. After initially depositing a layer of PAH for 30 min, PSS (1 g L−1) in 0.5 M NaCl was subsequently adsorbed in the same way. Adsorptions of PAH and PSS were performed five times in all at room temperature. After each adsorption step, excess polyelectrolyte was separated by centrifugation, and washed with ultrapure water several times.Adsorption of Au NPs
Au NPs were synthesized according to the method of Natan et al. with minor modifications.33 Typically, 4 mL of 1% sodium citrate was added to 400 mL 0.1 mM boiling HAuCl4 solution to prepare Au NPs. After 1 min boiling, the heat source was removed. The colloid solution was further stirred for 30 min and then stored in the dark at room temperature. Au NPs prepared had size of ∼30 nm. Negatively charged Au NPs were mixed with the obtained aqueous solution of UC microspheres for 6 h at room temperature. Excess Au NPs were separated by centrifugation, and washed with ultrapure water several times.Characterization
To demonstrate the overall uniformity and morphology of the particles, the samples were examined using scanning electron microscopy (SEM), using a JEOL SM-6360LV microscope equipped with an energy dispersive X-ray analyzer (EDX). Transmission electron microscopy (TEM) images were taken with a JEOL 2011 microscope (Japan) operating at 200 kV. High resolution transmission electron microscopy (HRTEM) was performed on a JEOL JEM 2100F instrument using an acceleration voltage of 200 kV to characterize composites. All samples were placed on a carbon-coated copper grid and dried at room temperature overnight. The crystalline structure was investigated by X-ray power diffraction (RIGAK, D/MAX 2550 VB/PC, Japan). Adsorption–desorption measurements were conducted on a Micromeritics ASAP 2010 apparatus at 77 K using nitrogen as the adsorption gas. The specific surface areas were calculated by the Brunauer–Emmett–Teller (BET) method. And the pore size distributions were calculated by the Barrett–Joyner–Halenda (BJH) method. Photoluminescence spectra were measured on a Fluorolog-3-P UV-VIS-NIR fluorescence spectrophotometer (Jobin Yvon, France), with a CW NIR laser at λ = 980 nm as the excitation source.Results and discussion
Morphologies of mesostructured Y2O3:Er@Au composites
The mesostructured Y2O3:Er@Au composites were prepared via integrating mesostructured Y2O3:Er microspheres with Au NPs, in which a multilayer polyelectrolyte served as spacer to avoid fluorescence resonant energy transfer (FRET) between the Au NPs and Y2O3:Er microspheres. SEM revealed the morphologies of the as-prepared mesostructured Y2O3:Er microspheres as shown in Fig. 2a. It is observed that the samples consist of monodisperse spheres with a mean particle size of 560 nm and the surfaces of the Y2O3:Er structures are rough. These particles are non-aggregated with narrow size distribution. The XRD patterns shown in Fig. 2b reveal that pure Y2O3:Er microspheres and Y2O3:Er@Au composites are formed. It is obvious that the well crystallized Y2O3:Er microspheres are cubic phase Y2O3 crystal (space group: Ia![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) ). All the strong peaks can be indexed to the data in the standard card (JCPDS files No.65-3178), with no peaks associate with impurities. In the case of Y2O3:Er@Au composites, the peaks of Y2O3:Er are observed in the composites, revealing that the surface-modified Au NPs do not change their phases, and four additional diffraction peaks of Au are also observed (JCPDS card No. 04-0784). This indicates that Au NPs do exist in the Y2O3:Er@Au composites. Fig. 2c represents the size distribution histogram of the microparticles, which confirms that the average diameter of the majority of the individual particles is about 560 nm. The corresponding EDX spectra (Fig. 2d) were taken at a number of selected positions of the sample and the elemental signatures of Y, Er, and O are essentially identical within experimental accuracy. In addition, there is still a little Si and Na, because SiO2 is not entirely removed by NaOH.
). All the strong peaks can be indexed to the data in the standard card (JCPDS files No.65-3178), with no peaks associate with impurities. In the case of Y2O3:Er@Au composites, the peaks of Y2O3:Er are observed in the composites, revealing that the surface-modified Au NPs do not change their phases, and four additional diffraction peaks of Au are also observed (JCPDS card No. 04-0784). This indicates that Au NPs do exist in the Y2O3:Er@Au composites. Fig. 2c represents the size distribution histogram of the microparticles, which confirms that the average diameter of the majority of the individual particles is about 560 nm. The corresponding EDX spectra (Fig. 2d) were taken at a number of selected positions of the sample and the elemental signatures of Y, Er, and O are essentially identical within experimental accuracy. In addition, there is still a little Si and Na, because SiO2 is not entirely removed by NaOH.
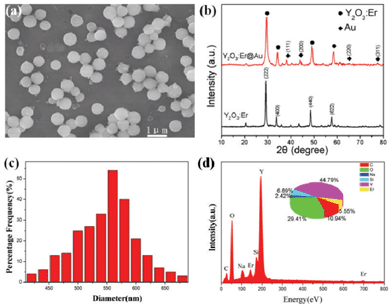 | ||
| Fig. 2 (a) SEM image of Y2O3:Er microspheres, (b) XRD patterns of Y2O3:Er microspheres and Y2O3:Er@Au composites, (c) histogram of the corresponding diameter distribution and (d) EDX spectrum of as-prepared mesostructured Y2O3:Er microspheres; quantified atomic elemental composition is given in the inset as a pie chart. | ||
A typical TEM image of mesostructured Y2O3:Er microspheres is shown in Fig. 3a and further confirms the uniform size of the sample. In Fig. 3b, a high-resolution TEM (HRTEM) image obtained from part of a Y2O3:Er microsphere shows a typical crystalline domain with an interplanar spacing of about 0.306 nm, comparable to the values of {222} of a cubic phase Y2O3 crystal. After the surface is modified with multilayer polyelectrolyte, Y2O3:Er microspheres show strong binding to Au NPs due to the strong electrostatic attraction between positively charged PAH coated Y2O3:Er microspheres and negatively charged Au NPs. When they are mixed in solution, small Au NPs are swiftly attached to the surfaces of Y2O3:Er microspheres to form core–shell structured Y2O3:Er@Au hybrid composites as shown in Fig. 3c; the inset image provides further evidence for the successful attachment of Au NPs with an average diameter of ∼30 nm. And the fast Fourier transform (FFT) pattern of the highlighted area reveals a regular hexagonal spot array, which is consistent with a highly crystalline particle and the measurement of lattice fringes shows the lattice spacing of 0.285 nm, characteristic of the Au plane (JCPDS files No. 04-0784) (Fig. 3d, inset).34
 | ||
| Fig. 3 TEM images of Y2O3:Er microspheres and Y2O3:Er@Au composites. (a) TEM image of Y2O3:Er microspheres, (b) HRTEM image of the Y2O3:Er microspheres, (c) TEM image of Y2O3:Er@Au microspheres and inset is a HRTEM image, (d) HRTEM image of Au NPs and the inset image corresponds to the FFT of the selected area outlined in red. | ||
In addition, the adsorption spectrum of the Y2O3:Er@Au composites is compared with that of the Y2O3:Er microspheres (Fig. 4a). A broad adsorption band peaked at ≈540 nm in the spectrum of Y2O3:Er@Au composites indicates the presence of Au NPs. All the results of TEM, XRD and adsorption spectra analyses indicate that Au NPs have been attached on the surfaces of Y2O3:Er microspheres.
 | ||
| Fig. 4 (a) UV-vis adsorption spectra of Y2O3:Er microspheres and Y2O3:Er@Au composites, (b) N2 adsorption/desorption isotherm for the as-prepared Y2O3:Er microspheres. | ||
To further investigate the specific surface area and porous nature of the Y2O3:Er microspheres, BET gas-sorption measurements were carried out. Fig. 4b shows the N2 adsorption/desorption isotherm and pore size distribution of an as-prepared Y2O3:Er sample. It can be seen that the Y2O3:Er microspheres show an N2 adsorption/desorption isotherm and the typical H3-hysteresis loop according to the IUPAC classification,35 which demonstrates the properties of typical mesoporous materials. The BET surface area of the sample is about 85.2 m2 g−1, the pore volume is 19.6 cm3 g−1 and the pore-size distribution reveals a narrow distribution apex centered at 5.7 nm (inset in Fig. 4b). This result further indicates that the as-prepared Y2O3:Er microspheres have porous structure.
UC luminescence properties
Fig. 5 presents the partial energy level diagrams of the Er3+ ions and the proposed mechanism to produce visible fluorescent radiation. The mechanisms of UC luminescence in Er3+ doped materials have been extensively studied.36,37 Through GSA (ground state adsorption), ESA (excited state adsorption) and ET (energy transfer) between two Er3+ ions and CET (cooperative energy transfer) between two Er3+ ions, mesostructured Y2O3:Er microspheres show UC fluorescence emissions at 525, 550 and 660 nm. | ||
| Fig. 5 Energy level diagrams of Er3+ ions as well as the proposed UC mechanisms for the green and red emissions. | ||
Fig. 6a shows the UC emission spectrum of an Er3+ doped Y2O3 sample under the excitation of 980 nm. In the UC luminescence spectrum, the red emission at 660 nm corresponds to the 4F9/2 → 4I15/2 transition of the Er3+ ions, and the green emission near 525 and 550 nm can be assigned to 2H11/2 → 4I15/2 and 4S3/2 → 4I15/2 transitions of Er3+ ions, respectively. To obtain a better understanding of the UC mechanism in Y2O3:Er and Y2O3:Er@Au, the UC emission intensity (I) was measured as a function of the laser power (P) (Fig. 6b). For the UC process, I is proportional to the nth power of P, that is:
| I ∝ Pn |
 | ||
| Fig. 6 (a) UC emission spectrum of a Y2O3:Er sample under 980 nm laser excitation. (b) Power dependence of the upconverted green emission (2H11/2 → 4I15/2) and red emission (4F9/2 → 4I15/2) of Y2O3:Er microspheres and Y2O3:Er@Au composites excited under 980 nm excitation. | ||
A plot of ln I versus ln P yields a straight line with slope n. The results are shown in Fig. 6b for the 4F9/2 → 4I15/2 red emission and 2H11/2 → 4I15/2 green emission. From Fig. 6b, the slopes n are 2.3 and 2.0 in Y2O3:Er samples, and 2.2 and 2.0 in Y2O3:Er@Au samples for red (4F9/2 → 4I15/2) and green (2H11/2 → 4I15/2) emissions under investigation, respectively. It was therefore determined that the green and red emission occurs via a two-photon process. Similarly, a two-proton UC mechanism is also involved to generate the UC emission in the Y2O3:Er@Au composites as presented in Fig. 6b. It is apparent that the attachment of Au NPs does not change the UC luminescence mechanism of the samples.
Enhancement of the UC luminescence
As we know, the UC green emission of Y2O3:Er matches well with the strong adsorption of Au NPs at λ ≈ 520 nm in the visible region.41 According to the theory of FRET, when the adsorption of the energy acceptor is close to the emission of the phosphor and when the donor and the acceptor are close enough, the emission of the energy donor (Y2O3:Er microspheres) will be quenched by the energy acceptor (Au NPs).42 To avoid FRET of UC luminescence, a multilayer polyelectrolyte is used as spacer by the LbL method, which is based on the electrostatic attraction between the oppositely charged species deposited, and its major advantage is that it permits the preparation of coated colloids of different shapes and sizes, with uniform layers of diverse composition as well as controllable thickness.43,44In order to verify the effectiveness of the self-assembly of PSS and PAH, the ζ-potential at different stages of surface modification of composite microspheres was measured. The prepared Y2O3:Er microspheres are highly positively charged because of the adsorption of hydronium ions,45 as shown in Fig. 7a, one layer of PSS changes the overall charge of a composite microsphere surface to negative, and the ζ-potential to decrease from ca. 35.1 mW to ca. −29.2 mW, which demonstrates successful PSS coating. The zeta potential of the microspheres alternates from positive to negative values with the alternating adsorption of the polyelectrolyte, which indicates that PAH and PSS are adsorbed alternately onto the surfaces of the microspheres. Finally, negatively charged Au NPs were adsorbed.
 | ||
| Fig. 7 (a) The zeta-potential vs. layer number of PSS and PAH on Y2O3:Er microspheres. (b) Normalized upconversion spectrum of Y2O3:Er microspheres (black line) and extinction spectrum of Au NPs (red line). | ||
All measurements were carried out with dried powder under 980 nm excitation from a laser; the corresponding multicolour UC emission spectra of Y2O3:Er and Y2O3:Er@Au were recorded under the same conditions, respectively, as shown in Fig. 8a. The spectrum of each displays sharp characteristic emission peaks. In the spectrum of Y2O3:Er@Au composites, enhancements of the multicolour UC emissions are achieved. Additionally, it is interesting to note that the maximum is positioned around 525 nm. The enhancement factors in the green region are much larger than those in the red region. Specifically, more than a 160% increase in emission intensity was observed at 525 nm and 550 nm, while an increase of 100% is seen at 660 nm.
 | ||
| Fig. 8 (a) UC spectra of Y2O3:Er microspheres and Y2O3:Er@Au microspheres under 980 nm excitation. (b) Enhancement factor after Au NPs attachment. | ||
The origin of the plasmonic enhancement effect from noble metal NPs has been attributed to two possible reasons in previous papers: (1) an increase in the effective excitation induced by local field enhancement (LFE); (2) an increase in both the nonradiative and radiative decay rate of fluorophores due to surface plasmon-coupled emission (SPCE), which is an enhancement of emission efficiency because of the coupling of the upconversion emission with the NP plasmonic resonance.46,47Fig. 7b shows that the plasmon resonance frequency of Au NPs overlaps well with the two major emission bands of the composites (525 nm and 550 nm) so that SPCE can occur, which can thus increase the radiative decay rate, emission efficiency, and intensity of the composites. With a better plasmonic coupling near the plasmon resonance frequency, the SPCE is also a reason why the observed enhancement factor is larger for green emission than for red emission (Fig. 8b). These studies suggest that SPCE plays an important role in the spectral dependent enhancement of upconversion emission, although other effects such as LFE may also contribute.
Conclusions
In summary, mesostructured Y2O3:Er microspheres have been successfully prepared via using MS spheres as a hard template. A binary assembly of mesostructured Y2O3:Er microspheres and Au NPs was also developed through facile LbL technology. We further demonstrated that this hybrid composite achieved enhanced multicolour UC emission and this enhancement can be at least partly attributed to SPCE, which can increase the radiative decay rate and emission efficiency and further studies will be necessary to fully elucidate the exact underlying mechanism. This work provides a novel hybrid structure for applications such as bioimaging, bioassays and detection.Acknowledgements
We thank the National Natural Science Foundation of China (20925621, 20976054, and 21176083), the Special Projects for Nanotechnology of Shanghai (11nm0500800) the Fundamental Research Funds for the Central Universities (WD1013015 and WD1114005), and the Program for Changjiang Scholars and Innovative Research Team in University (IRT0825), and the Shanghai Leading Academic Discipline Project (project number: B502) for financial support.References
- H. Schäfer and M. Haase, Angew. Chem., Int. Ed., 2011, 50, 5808 CrossRef.
- C. Li and J. Lin, J. Mater. Chem., 2010, 20, 6831 RSC.
- Z. Xu, P. Ma, C. Li, Z. Hou, X. Zhai, S. Huang and J. Lin, Biomaterials, 2011, 32, 4161 CrossRef CAS.
- F. Wang and X. G. Liu, Chem. Soc. Rev., 2009, 38, 976 RSC.
- H. P. Paudel, L. L. Zhong, K. M. Bayat, F. Baroughi, S. Smith, C. Lin, C. Y. Jiang, M. T. Berry and P. S. May, J. Phys. Chem. C, 2011, 115, 19028 CAS.
- Z. Xu, C. Li, P. Ma, Z. Hou, D. Yang, X. Kang and J. Lin, Nanoscale, 2011, 3, 661 RSC.
- S. Gai, P. Yang, C. Li, W. Wang, Y. Dai, N. Niu and J. Lin, Adv. Funct. Mater., 2010, 20, 1166 CrossRef CAS.
- A. J. Haes and R. P. Van Duyne, Anal. Bioanal. Chem., 2004, 379, 920 CrossRef CAS.
- S. Eustis and M. A. El-Sayed, Chem. Soc. Rev., 2006, 35, 209 RSC.
- W. L. Barnes, A. Dereux and T. W. Ebbesen, Nature, 2003, 424, 824 CrossRef CAS.
- O. Kedem, A. B. Tesler, A. Vaskevich and I. Rubinstein, ACS Nano, 2011, 5, 748 CrossRef CAS.
- F. Le, D.W. Brandl, Y. A. Urzhumov, H. Wang, J. Kundu, N. J. Halas, J. Aizpurua and P. Nordlander, ACS Nano, 2008, 2, 707 CrossRef CAS.
- K. G. Thomas and P. V. Kamat, Acc. Chem. Res., 2003, 36, 888 CrossRef CAS.
- J. Zhang, Y. Fu, M. H. Chowdhury and J. R. Lakowicz, Nano Lett., 2007, 7, 2101 CrossRef CAS.
- F. Zhang, G. B. Braun, Y. F. Shi, Y. C. Zhang, X. H. Sun, N. O. Reich, D. Y. Zhao and G. Stucky, J. Am. Chem. Soc., 2010, 132, 2850 CrossRef CAS.
- K. S. Kim, J. H. Kim, H. Kim, F. Laquai, E. Arifin, J. K. Lee, S. Yoo and B. H. Sohn, ACS Nano, 2012, 6, 5051 CrossRef CAS.
- A. J. Amali, P. Saravanan and R. K. Rana, Angew. Chem., Int. Ed., 2011, 50, 1318 CrossRef CAS.
- G. Decher and G. Schneider, Nano Lett., 2006, 6, 530 CrossRef.
- M. Li, X. F. Yu, S. Liang, X. N. Peng, Z. J. Yang, Y. L. Wang and Q. Q. Wang, Adv. Funct. Mater., 2011, 21, 1788 CrossRef CAS.
- N. Liu, B. S. Prall and V. Klimov, J. Am. Chem. Soc., 2006, 128, 15362 CrossRef CAS.
- J. L. West and N. L. Halas, Annu. Rev. Biomed. Eng., 2003, 5, 285 CrossRef CAS.
- J. H. Song, T. Atay, S. Shi, H. Urabe and A. V. Nurmikko, Nano Lett., 2005, 5, 1557 CrossRef CAS.
- W. Feng, L. D. Sun and C. H. Yan, Chem. Commun., 2009, 4393 RSC.
- S. Schietinger, T. Aichele, H. Q. Wang, T. Nann and O. Benson, Nano Lett., 2010, 10, 134 CrossRef CAS.
- H. Zhang, Y. J. Li, I. A. Ivanov, Y. Q. Qu, Y. Huang and X. F. Duan, Angew. Chem., Int. Ed., 2010, 49, 2865 CrossRef CAS.
- N. Liu, W. P. Qin, G. S. Qin, T. Jiang and D. Zhao, Chem. Commun., 2011, 47, 7671 RSC.
- Z. Q. Li, L. M. Wang, Z. Y. Wang, X. H. Liu and Y. J. Xiong, J. Phys. Chem. C, 2011, 115, 3291 CAS.
- Z. J. Wang, L. N. Wu, H. J. Liang, W. Cai, Z. G. Zhang and Z. H. Jiang, J. Alloys Compd., 2011, 509, 9144 CrossRef CAS.
- J. Guzman and A. Corma, Chem. Commun., 2005, 743 RSC.
- S. Wang, F. Gu, C. Li and M. Lü, Cryst. Growth Des., 2007, 7, 2670 CAS.
- Y. X. Li, Y. H. Zhu, C. Y. Li, X. L. Yang and C. Z. Li, Mater. Lett., 2009, 63, 1068 CrossRef CAS.
- J. Zong, Y. Zhu, X. Yang and C. Li, Mater. Sci. Eng., C, 2011, 31, 166 CrossRef CAS.
- K. C. Grabar, R. G. Freeman, M. B. Hommer and M. J. Natan, Anal. Chem., 1995, 67, 735 CrossRef CAS.
- N. Ortiz and S. E. Skrabalak, Cryst. Growth Des., 2011, 11, 3545 CAS.
- C. M. Zhang, Z. Y. Hou, R. Chai, Z. Y. Cheng, Z. H. Xu, C. X. Li, L. Huang and J. Lin, J. Phys. Chem. C, 2010, 114, 6928 CAS.
- J. Zhang, S. W. Wang, T. J. Rong and L. D. Chen, J. Am. Ceram. Soc., 2004, 87, 1072 CrossRef CAS.
- G. S. Yi, B. Q. Sun, F. Z. Yang, D. P. Chen, Y. X. Zhou and J. Cheng, Chem. Mater., 2002, 14, 2910 CrossRef CAS.
- X. Bai, H. W. Song, G. H. Pan, Y. Q. Lei, T. Wang, X. G. Ren, S. Z. Lu, B. Dong, Q. Dai and L. Fan, J. Phys. Chem. C, 2007, 111, 13611 CAS.
- Y. Sun, H. Liu, X. Wang, X. Kong and H. Zhang, Chem. Mater., 2006, 18, 2726 CrossRef CAS.
- H. Guo, N. Dong, M. Yin, W. Zhang, L. Lou and S. Xia, J. Phys. Chem. B, 2004, 108, 19205 CrossRef CAS.
- T. A. Taton, C. A. Mirkin and R. L. Letsinger, Science, 2000, 289, 1757 CrossRef CAS.
- L. Y. Wang, R. X. Yan, Z. Y. Huo, L. Wang, J. H. Zeng, J. Bao, X. Wang, Q. Peng and Y. D. Li, Angew. Chem., Int. Ed., 2005, 44, 6054 CrossRef CAS.
- G. Decher, Science, 1997, 277, 1232 CrossRef CAS.
- D. Wang, A. L. Rogach and F. Caruso, Nano Lett., 2002, 2, 857 CrossRef CAS.
- H. X. Zhong, Y. L. Ma, X. F. Cao, X. T. Chen and Z. L. Xue, J. Phys. Chem. C, 2009, 113, 3461 CAS.
- T. Jiang, Y. Liu, S. S. Liu and W. P. Qin, J. Alloys Compd., 2012, 377, 81 CAS.
- S. Schietinger, T. Aichele, H. Q. Wang, T. Nann and O. Benson, Nano Lett., 2010, 10, 134 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |